
ABSTRACT
Standardised mortality rates for liver disease in the UK have increased 400% since 1970. However, evidence from a large number of animal models and clinical trials indicates that liver fibrosis and even cirrhosis are potentially reversible if the underlying cause can be successfully removed. Nevertheless, in a significant number of patients cure of the underlying disease may not result in fibrosis regression, and no antifibrotic drug has been licenced by the United States Food and Drug Administration or the European Medicines Agency. Dissection of the mechanistic pathways and regulatory factors that characterise matrix remodelling and architectural repair during fibrosis regression are revealing novel therapeutic approaches to induce liver repair. Points of attack in the fibrotic cascade include promoting the loss of hepatic myofibroblasts, inhibiting profibrogenic properties of myofibroblasts, stimulating degradation of accumulated liver scar tissue, targeting the immune response, and cell-based therapies. Therapeutic candidates are now being evaluated in early-phase human trials but translation into the clinic will require careful patient selection and stratification, and the definition and validation of clinically meaningful endpoints.
Introduction
Liver disease is the fifth largest cause of death in the UK. The average age of death from liver disease is 59 years, compared to 82–84 years for heart and lung disease or stroke. A recent cohort study has shown that between 1998 and 2009 the crude incidence of liver cirrhosis increased by 50.6%.1 The percentage change in incidence of cirrhosis between 1998 and 2009 for both men (52.4%) and women (38.3%) was greater than that seen for the top four most commonly diagnosed cancers in the UK (breast, lung, bowel and prostate).
Non-alcoholic fatty liver disease (NAFLD) is now the commonest liver disease in both rich and poor countries, and affects public health and healthcare globally. It is estimated that NAFLD will increase direct and indirect medical costs by 26% over the next five years. Of the 25% of the UK population now categorised as obese, most will have NAFLD and approximately 10% of these people have been diagnosed in community studies to have evidence of advanced liver fibrosis that leads to cirrhosis.2 Of those patients with cirrhosis, 5–10% will develop hepatocellular carcinoma (HCC). Type-2 diabetes, which is increasing in frequency in parallel with obesity, is strongly associated with NAFLD. According to Duncan Selbie, head of Public Health England, the obesity crisis could result in the number of people with type-2 diabetes trebling during the next 20 years to 6.2 million by 2034.3
Fibrosis, or scarring, of the liver is a generic wound healing response to chronic liver disease regardless of aetiology.4 Progressive fibrosis eventually evolves to cirrhosis, with fibrotic bands, regenerative nodules and vascular distortion leading to liver cell dysfunction, portal hypertension (PHT), HCC and premature death (Fig 1). Liver fibrosis is a clinically silent process, such that many patients present at an advanced stage when disease-specific therapy has limited impact. Central to this is the recognition that cirrhosis is not simply severe fibrosis, but a more complex pathological condition with reversible and irreversible components. Detailed analysis of the cellular and molecular mechanisms that mediate liver fibrosis has provided a framework for therapeutic approaches to prevent, slow or even reverse fibrosis or cirrhosis.6 The goal of such treatments would be stasis or regression of fibrosis to pre-cirrhotic stages to prevent the development of liver failure, HCC and/or reduce PHT and its complications, such as variceal haemorrhage, hepatorenal syndrome and encephalopathy (Fig 1). Despite this rationale, no antifibrotic drugs are currently licensed for use in human liver fibrosis. With epidemiological projections for the future prevalence of cirrhosis painting an increasingly gloomy picture and a shortfall in donors for liver transplantation, the clinical urgency for new therapies is high and there is increasing interest from stakeholders keen to exploit the market potential for antifibrotics. Once treatments are identified, we need reliable non-invasive techniques to measure changes in fibrosis over time, as liver biopsy is invasive, and ethically and practically unsuitable for repetitive use in the context of clinical trials.
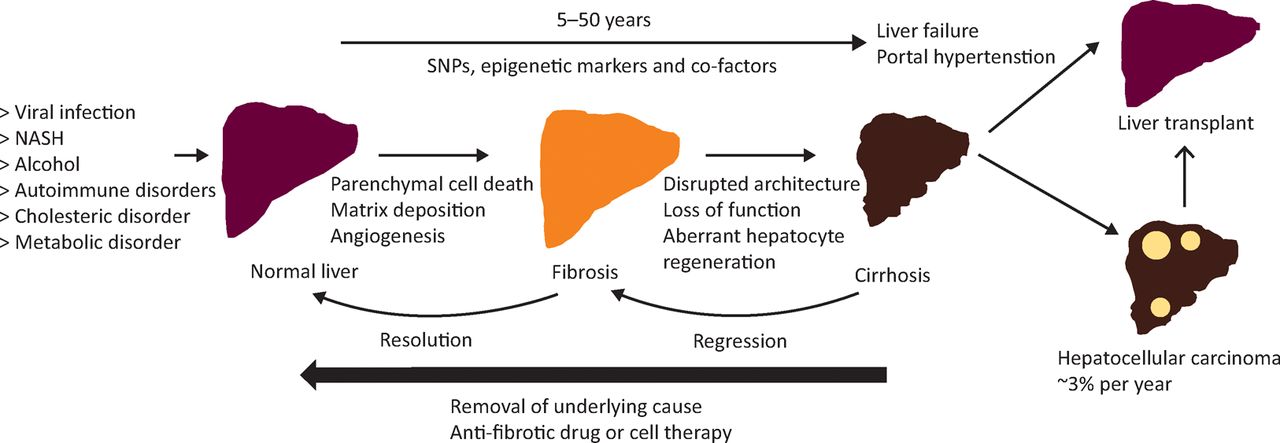
Natural history of liver fibrosis. Fibrosis is the liver's wound healing response to many causes of chronic injury. Once cirrhosis is established the potential for reversing this process is decreased and complications develop. Genetic polymorphisms, epigenetic marks and co-factors may modulate the risk of fibrosis progression. If the cause of fibrosis is removed, resolution of early hepatic fibrosis can occur. Even in cirrhosis, regression of fibrosis improves clinical outcomes. Antifibrotic therapies are emerging that could slow, halt or reverse fibrosis avoiding the need for liver transplantation. HCC is rising in incidence worldwide and is a major cause of liver-related death in patients with cirrhosis. HCC = hepatocellular carcinoma; NASH = non-alcoholic fatty liver disease; SNP = single nucleotide polymorphism. Adapted with permission.5
Evidence for reversibility of fibrosis
Spontaneous resolution of liver fibrosis is a well established phenomenon in a range of rodent and clinical models of diverse aetiology.7 Indeed, relatively large scale trials in chronic viral hepatitis have shown very encouraging results in terms of remodelling of fibrosis and even cirrhosis.8 Critically, regression of fibrosis also appears to correlate with improved clinical outcomes. For example, in patients with hepatitis C (HCV)-related cirrhosis treated with antivirals, ‘regressors’ had better 10-year survival rates than ‘non-regressors’ (100 vs 74%).9 By contrast, patients in the HALT-C trial who exhibited faster fibrosis progression (26.7% of HALT-C patients) had increased clinical outcomes (46 vs 8%, p<0.0001) and HCC (10% vs 3%, p=0.006) over 7-year follow up.10 Additionally, patients with HCV-related cirrhosis who achieved sustained virological response and regression of fibrosis with pegylated-interferon/ribavirin also had reduction in PHT (hepatic venous pressure gradient).11
Liver fibrogenesis
The principal cell type responsible for scar deposition in the damaged liver is the hepatic stellate cell (HSC), which transdifferentiates into a myofibroblast (MF) phenotype following liver injury. In addition to extracellular matrix (ECM) synthesis, activated HSC-MFs have a range of additional pro-fibrogenic properties, including immunomodulatory functions.12 Furthermore, hepatic MFs are the principal source of tissue inhibitors of metalloproteinases (TIMPs), in particular TIMP1.13 TIMPs are secreted into the extracellular milieu and inhibit the group of matrix-degrading enzymes known as matrix metalloproteinases (MMPs), which are potentially capable of breaking down a variety of scar tissue components including fibrillar collagens. Importantly, even during progressive hepatic fibrosis, a range of MMPs are expressed in the liver, demonstrating the inherent capacity for ECM degradation. However, TIMP expression by MFs inhibits this latent hepatic MMP activity, preventing scar degradation and producing a microenvironment favouring ECM deposition.14
Key mechanisms of fibrosis regression
For fibrosis regression to occur there must be cessation of active scar production, and also degradation of the fibrotic tissue that has already been deposited in order to restore normal tissue architecture. Understanding the key cellular and molecular processes that promote fibrosis regression is critical for the identification of new mechanistic strategies for tackling liver fibrosis. Seminal studies in animal disease models have shown that, after the insult that induced fibrosis is withdrawn, TIMP levels decrease dramatically, MMP activity increases and scar is degraded.15 A critical observation in these studies was the loss of MFs from the receding hepatic scar. As well as removing the principal source of scar ECM from the liver, the decline in MF numbers also leads to a critical reduction in levels of TIMPs. The mechanisms by which hepatic MFs are lost from the liver include apoptosis16 (programmed cell death), senescence17 (a stable form of cell cycle arrest) and also reversion to a more quiescent phenotype. Two recent landmark papers used transgenic mice which enabled lineage tracing of activated HSC-MFs and demonstrated that around 40% of these cells reverted to a quiescent phenotype following cessation of liver injury.18,19 Both studies also showed that reverted HSCs were not identical to pre-injury quiescent HSCs, remaining in a ‘primed’ state characterised by an augmented response to further fibrogenic stimuli – an observation that may have implications for human liver fibrosis, where repetitive cycles of injury/recovery (eg in alcohol-related liver disease) are common.
Hepatic macrophages have re-emerged as key regulators of matrix remodelling (Fig 2), with studies demonstrating a capacity for injury-inducing or repair-promoting roles in liver fibrosis.20,21 Our group recently identified a population of Ly-6Clo ‘restorative macrophages’, derived from a phenotypic switch of pro-fibrogenic Ly-6Chi macrophages (potentially induced by macrophage phagocytosis), which was responsible for murine liver fibrosis resolution following chronic CCl4 injury.21 What remains to be seen is how closely these observations about murine macrophage heterogeneity relate to human disease.
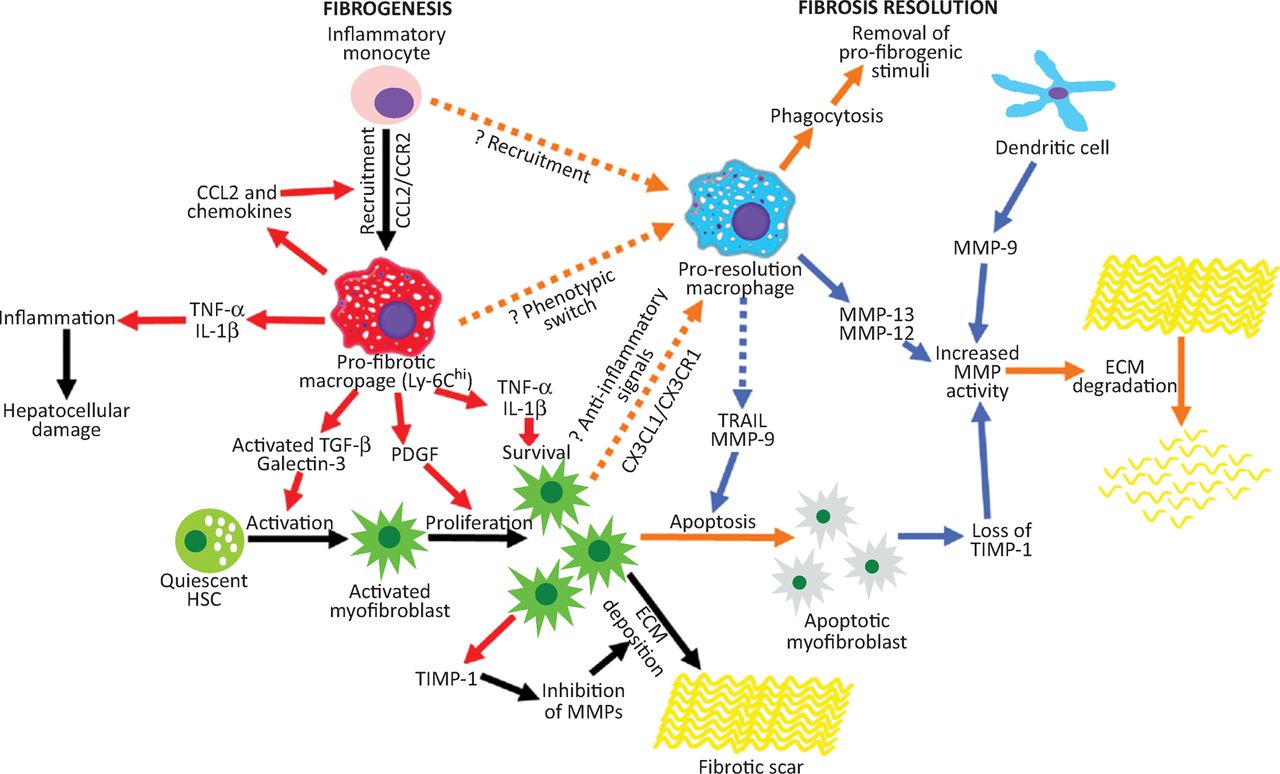
The role of macrophages in liver fibrogenesis and fibrosis regression. Inflammatory Ly-6Chi monocytes are recruited to the injured liver and form the population of profibrotic hepatic macrophages which release proinflammatory cytokines/chemokines that promote hepatocellular damage and MF activation, proliferation and survival. These profibrogenic macrophages change phenotype in response to phagocytosis which results in generation of Ly-6Clo restorative hepatic macrophages that mediate fibrosis regression by clearing cellular debris, downregulating proinflammatory cytokines, producing proapoptotic factors for MFs, upregulating MMPs, and expressing growth factors that promote hepatocyte regeneration. VEGF has a dual role, promoting monocyte recruitment but also proresolution effects in hepatic macrophages. Adapted with permission.4CCL = chemokine (C-C motif) ligand; CCR = C-C chemokine receptor type; CXCL = chemokine (C-X-C motif) ligand; CXCR = chemokine (C-X-C motif) receptor; ECM = extracellular matrix; HSC = hepatic stellate cell; IL = interleukin; MF = myofibroblast; MMP = matrix metalloproteinases; PDGF = Platelet-derived growth factor; TGF = transforming growth factor; TIMP = tissue inhibitors of metalloproteinases; TNF = tumour necrosis factor; VEGF = vascular endothelial growth factor.
Factors limiting reversibility of hepatic fibrosis
Persistent liver injury leads to pathological alterations in the quantity and composition of the ECM (particularly an increase in fibrillar collagens) as well as biochemical modifications that render the fibrotic liver less susceptible to remodelling and repair. Mature scar ECM, composed of cross-linked collagen and elastin,22 is more resistant to proteases, and fibrils sequestered in deeper portions of scar are inaccessible to degrading enzymes. The pattern/distribution of fibrosis also differs with aetiology and is associated with varying degrees of angiogenesis and potential for reversibility. In addition, acellular scars lack endogenous sources of MMPs (inflammatory cells and MFs) and angio-architectural changes, such as vascularised septa that accompany cirrhosis, are probably irreversible and as such represent a point-of-no-return.23
Therapeutic targets in liver fibrosis
The best antifibrotic treatment is to remove the underlying cause24 but in many patients this is simply not possible. Consequently, fibrosis researchers and (increasingly) the pharmaceutical industry have focused on the development of antifibrotic treatments. A vast number of putative antifibrotic interventions have emerged from preclinical research in recent years.6 These include medicinal compounds, dietary measures and cellular therapies. A summary of mechanistic strategies to induce reversal of hepatic fibrosis is shown in Box 1. Points of attack in the fibrotic cascade include: promoting apoptosis, senescence or reversion of hepatic MFs, inhibiting specific properties of MFs (eg fibrogenesis), stimulating degradation of accumulated scar ECM, and targeting the immune response. To date, rodent models have not been reliable in terms of predicting clinical efficacy. Biological differences in rodent and human liver fibrosis may underpin this disconnect. Combinations of targeted antifibrotic therapies that inhibit fibrogenesis, induce fibrolysis or influence different cell types could theoretically be highly effective, but multiagent approaches have not yet been extensively tested in preclinical studies. The concept of ‘core’ versus ‘regulatory’ pathways has been proposed to identify the most promising targets for antifibrotic drug discovery/design.25 Examples of probable core pathway constituents include lysyl oxidase-like 2 (LOXL2),26 αv integrins27 and transforming growth factor beta (TGF-β).28 These molecules have been shown in animal models to be critical to fibrosis in more than one tissue and antifibrotic medicines targeting LOXL2 and TGF-β signalling are being evaluated in clinical trials. Cell-specific carrier approaches, such as coupling of compounds to albumin or incorporation in liposomes,28 may achieve effective delivery of potent drugs and biological agents (eg cytokines, siRNAs) to the key target cells in liver fibrosis – HSC-MFs and hepatic macrophages. Therapeutic administration of cells (eg macrophages or stem cells) has been shown to induce both antifibrotic and proregenerative effects in vivo. There are some concerns regarding the use of autologous unsorted bone marrow cell (BMC) infusions for the treatment of liver fibrosis, as BMCs contain mesenchymal stem cells that can differentiate into MFs in certain settings. A better strategy might be to use macrophages, haematopoetic stem cells or BM mononuclear cells. In particular, macrophages are biologically well defined, and can be generated in large numbers from CD14+ blood monocytes after isolation and differentiation. Injected autologous macrophages have been shown to be antifibrotic and anti-inflammatory, and stimulate liver regeneration in models of liver injury and fibrosis.29 Liver-engrafted macrophages, through secretion of chemokines, can also recruit endogenous cells to the scar to amplify their effect.
Potential mechanistic strategies for tackling liver fibrosis.
Translational challenges
A number of important hurdles remain with respect to clinical translation of emerging antifibrotic therapies.30 Firstly, the optimal preclinical models for establishing proof of concept must be defined and standardised,31 in order to avoid future disappointment in human trials. In addition, antifibrotic drug development is currently hindered by the long and usually asymptomatic natural history of most fibrotic liver diseases, varying clinical phenotypes (likely influenced by a number of genetic and environmental risk factors that have not yet been defined), lack of validated endpoints (both for cirrhotic and non-cirrhotic trial populations), and lack of data to support the Food and Drug Administration's ‘reasonably likely to predict clinical benefit’ standard of surrogate endpoints. Although trial populations with more advanced stage disease have higher clinical event rates (approximately 4% per year), such hard endpoints may not be suitable for some antifibrotic drug targets (eg metabolic therapies in non-alcoholic steatohepatitis). In the future, careful selection and stratification of trial participants will likely be based on those most likely to progress/respond. Indeed, fibrosis in patients with established cirrhosis is inherently less reversible. Until these issues are overcome, translation (and prioritisation) of antifibrotic drug candidates will be challenging.
- © Royal College of Physicians 2015. All rights reserved.
References








{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.