
Sir John Bell opened his talk with a history of the classification of disease and the contributions of Koch, Erlich, Pasteur, Garrod, Virchow and Osler in particular. He continued his lecture with a discussion on the importance of genetics.
Genes and environment together affect pathways involved in disease and in turn lead to many downstream alterations that reflect these integrated effects. mRNA and protein signatures of disease have, in some circumstances, proved valuable as signatures of the interaction of genes and environment (systemic lupus erythematosus (SLE) and breast cancer).1 The signatures integrate the effects of the genetic and environmental events initiating disease, but such signatures also risk identifying secondary or reactive events not involved in the primary disease pathway.
The march of genetic technologies
The central role of genetics in disease biology has made the discovery of the genetic factors associated with the major common diseases one of the most significant priorities of modern molecular science. The journey to uncover these disease determinants, which is not yet complete, has evolved over 20 years and provided many disappointments but, increasingly, also many successes. If diseases were to be classified by mechanisms, then these are the insights that are essential.
Long before molecular genetic tools became available, other methods for typing protein polymorphisms were used to identify genetic determinants of disease. The most significant and major genetic association with common complex disease emerged with the serological typing of HLA antigens that emerged from the work of Dausset, Bodmer, McDevitt and others.2 Maternal serum reactive with paternal HLA antigens through sensitisation by the fetus revealed a highly polymorphic series of antigens on most nucleated cells. The genes encoding these HLA antigens mapped to a region on chromosome 6p and are now recognised to be responsible for controlling much of the adaptive immune response. The association of particular HLA alleles with autoimmune diseases (famously HLA-B27 and ankylosing spondylitis) has been an exemplar for those mapping other genetic variants in common disease, exceptional only because of the large size of its effect. Components of the major histocompatibility complex have also been associated with liability to infectious disease and cancer. Most of the associations derived from serology have now been replicated and refined by molecular technologies. The high degree of polymorphism in these HLA genes has probably been driven by the powerful selective forces that determine the immune response to individual pathogens, providing a powerful evolutionary pressure to diversify. These genetic effects are models of how common polymorphisms, selected for important beneficial effects, can contribute to the susceptibility to common disease.
Biochemical genetics proved not to have the capacity to seriously address the broader genetic basis of common disease. This had to await the developments that allowed more direct and systematic characterisation of genomic DNA. This capability emerged half a century after Garrod and again involved sequential waves of technological innovation. Early work recognised significant variation between individuals, both large structural variations recognised by cytogenetics or single base-pair variation identified by restriction fragment length polymorphisms and Southern blots. However, these tools proved inadequate for efficiently analysing the genome. The recognition of repeat sequence variation in the genome provided a wider set of markers that could be used systematically, firstly through the use of mini satellite sequences described originally by Jeffreys et al3 and then the recognition of di- and tri-nucleotide repeats.4 These provided frequent polymorphic markers throughout the genome, sufficient to allow non-hypothesis based discovery of markers co-segregating with disease.
Genetic linkage was the approach initially taken for genome-wide analysis and immediately success was achieved in a wide range of Mendelian disorders. Cystic fibrosis, Huntington's disease, tuberous sclerosis, spinal muscle atrophy, myotonic dystrophy and now more than a thousand Mendelian single gene diseases, all proved tractable using families and linkage studies. This work validated the work of Garrod by demonstrating the precise inherited ‘chemical’ defects accounting for many Mendelian metabolic disorders, as he would have predicted.
During the 1980s and 1990s, these genomic tools were further developed to be applied to common complex diseases. Linkage analysis was undertaken in large pedigrees and genetic analysis was used to find markers that segregated more commonly into affected sibling pairs than one would expect by chance. Progress was made, and many regions of the genome apparently linked to disease were discovered, along with a few novel disease genes. These methods were constrained, however, as the genomic regions identified by linkage or affected sibling pair analysis were large and imprecisely defined, while the evidence for linkage was often weak and not reproducible. This made the characterisation of the precise variant responsible for disease difficult and, although this era had its successes, the wealth of information expected on genetics of common disease was not forthcoming.
Two major developments facilitated the wider identification of genes and pathways responsible for common disease. The first was the generation of the sequence of the human genome.5 This created the template that would allow the second advance, the mapping of genes and the identification by sequencing of the common single nucleotide variation (single nucleotide polymorphisms, SNPs) in the genome. The SNP Consortium was pivotal in identifying millions of variable markers that could be used to pinpoint disease genes.6 Coincidental with these developments, it had been argued that genome-wide association studies could provide a more powerful tool than linkage studies in searching for disease-related variants.7 When used in relation to candidate genes, association studies had proved to be remarkably unproductive, but the prospect of a set of genome-wide markers alongside a strategy for using them provided the opportunity for the field to progress rapidly.
The presence of large numbers of linkage disequilibrium (LD) blocks was essential for the whole genome association strategy to work. LD represents the association of polymorphisms at different sites along a haplotype occurring more frequently than would have been expected had they segregated independently. LD can be attributed to founder effects, regions where recombination is suppressed or where variants are co-selected because they interact functionally. The HLA region had provided strong evidence of LD, but only when the large numbers of SNPs identified by resequencing were available could the patterns of LD across the whole genome be defined. The HapMap project8,9 allowed these regions of LD to be resolved and this set the stage for the selection of a subset of SNP defining most of the regions of the genome through LD blocks. Array technology, developed originally by Ed Southern, could then be used to scan whole genomes for association with disease compared to controls. The stage was set for another chapter in the discovery of genetic factors in disease.
The first successful genome-wide association study (GWAS) using large numbers of SNPs typed in cases and controls using arrays was of macular degeneration.10 This study identified that complement Factor H was strongly associated with this disease, introducing a totally novel concept that an inflammatory process was involved in the pathogenesis of this disease. Large numbers of patient samples had already been accumulated in many other disorders and immediately the race began to characterise genetic determinants in a wide range of common complex diseases. Globally, many centres were involved and amongst the most influential of the early studies was the Wellcome Trust Case Control Consortium (WTCCC).11 In 2006, this programme, chaired by Peter Donnelly, set about undertaking large GWAS studies in seven different diseases. A crucial question in such studies was the number of patient samples needed to detect significant associations, particularly given the use of multiple testing. The WTCCC proposed that 2,000 patients in a two-stage strategy would provide the power to detect disease susceptibility genes. The study was a remarkable success. Multiple loci were found in most of the diseases studied with many demonstrating significant associations. Replication in other studies soon illustrated that this approach produced fully reproducible results. For the first time, the factors that determined an individual's liability to common disease were being defined in large numbers.
Since 2007, more than 1,000 genetic variants associated with common diseases have been found and replicated using this approach in over 150 diseases. Meta-analysis using multiple studies has allowed the analysis of tens of thousands of patients with a single disease. These data have revealed both genes and pathways involved in disease pathogenesis and, while some would have been predicted based on our understanding of disease (eg IL-2R, PTPN in type 1 diabetes),12 most mechanisms identified had never previously been considered as having a role in disease. It is clear from the accumulated GWAS data that, after centuries of clinical observation and study, our understanding of disease pathogenesis for most diseases has been rudimentary. The discovery of even a portion of the genetic architecture of disease had a profound effect on our understanding of disease pathogenesis and will change our approach to defining and treating these diseases forever.
The impact of genetics on our understanding of both inflammatory and metabolic disease has been particularly impressive in providing new insights into the mechanisms of these diseases. Studies of inflammatory bowel disease (IBD) revealed the power of genetics to clarify disease pathways and also to illustrate how pathways can demonstrate important relationships between apparently distinct clinical diseases. The aetiology of IBD has been the subject of debate for decades. Clinically, the separation into two entities, ulcerative colitis and Crohn's disease (CD), based on clinical and pathological variations, is well recognised. Both diseases have multiple genetic susceptibility determinants with some shared genes or pathways. Crohn's disease has been one of the most interesting diseases to be characterised by genetics as over 50 susceptibility genes have been uncovered.13–15 It is not, however, the number of genes which is most interesting in this disease, but similarity between the pathways identified in CD and those seen in other disorders and the insights into pathogenesis we have derived from these data that demonstrate how powerful the genetic approach can be.
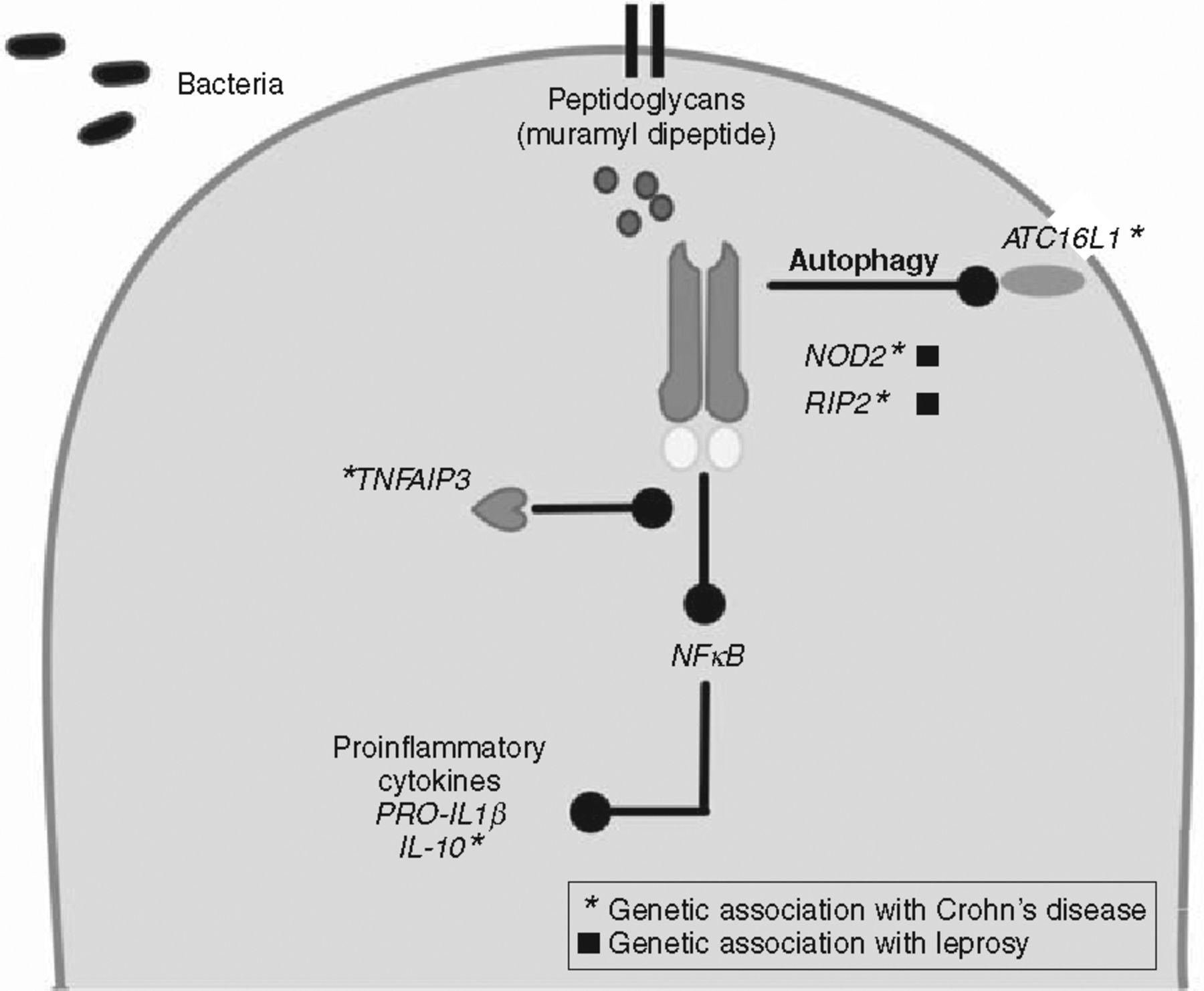
Two of the key pathways highlighted by genetic studies in CD use the inflammasome – involved in activating innate immune responses – and the IL-23 receptor pathway. Both these pathways lead to transcriptional activation amplifying the pro-inflammatory response through production of cytokines. The identification of NOD-2 by linkage studies provided the first link to inflammasome activity in CD and subsequent associated genes ATG16L1, RIPK2, NLRPS and IRGM are all implicated in CD and inflammasome function.16–19 Further, studies have also implicated IL-18 and TNFAIP3 as inflammatory mediators functionally linked to this complex. NOD-2 appears to be pivotal both in NF-kB signalling and also in recruiting ATG16L1 to induce autophagy independently of transcriptional activation. Autophagy is the cellular process of response to intracellular microbes. Variants associated with CD cells, including those at NOD-2 and related proteins (ATG16L1), disable the cell's ability to handle bacteria appropriately through the autophagy pathway.20–22 In addition, NOD-2 and ATG16L1 variants also independently disable the ability of dendritic cells to process and present bacterial antigens appropriately.20 Mouse models have shown the importance of pathogenic viruses and commensal bacteria in creating the aberrant inflammation in the gut in the presence of mutated NOD-2.23 The inflammasome and autophagy pathways are clearly novel and important components of CD aetiology.
Interestingly, many of these CD susceptibility genes have also emerged in studies of genetic susceptibility to the mycobacterial disease, leprosy.24,25 NOD2, RIP2K, and TNFSF15 have emerged from these genetic studies as does IL-12B, another cytokine associated with Crohn's disease. Both diseases appear to share a set of genetic determinants that affect the inflammosome and its role in the early innate response to bacterial antigens (Fig 1). This linkage indicates that determinants of CD susceptibility may have emerged from selection originally driven by leprosy. The similarities between mycobacterial infection and CD have been widely recognised pathologically and have suggested a role for atypical mycobacterium in the disease. It may prove that activation of the same pathways accounts for shared pathological features.

Multiple genetic factors define inflammasome pathways in Crohn's disease and leprosy.
CD also is associated with multiple components of the IL-23R pathway.26 Associations have been discovered with the IL-23 receptor, IL-12RB, STAT and JAK2, all part of the same pathway known to stimulate a unique T cell population, Th17 cells,27,28 recognised to be associated with autoimmune inflammatory responses. Variants in this IL-23 pathway are, however, also strongly shared by another autoimmune disease, ankylosing spondylitis (AS) where Th17 populations also play an important role.29,30,31 In AS, IL-23R, JAK2 and STAT have all been genetically associated with disease as they are with CD.32 These diseases can, on occasion, coexist and AS patients are recognised often to have asymptomatic terminal ileitis. Although HLA-B27 dominated genetic thinking about AS for many years, IL-23R and two genes contributing to its signalling pathway, JAK2 and STAT3, are also important in this disease and clearly implicate this pathway and Th17 cells in AS.
In mechanistic terms, therefore, it appears that CD has an important overlap with AS – considerably more than it has with ulcerative colitis despite the inflammation targeting different organs. These and other data allow us to begin to recognise several distinct types of autoimmunity. CD, AS and psoriasis have significant mechanistic overlap and share little with diseases such as type 1 diabetes, MS and coeliac disease. Most striking, however, is that all these insights into pathogenic mechanisms in CD have emerged from genetics – none were suspected when phenotype alone was relied on for insights into aetiology.
Diabetes represents another excellent example of how the developments in human molecular genetics have transformed our understanding of the pathogenesis of disease and provides an opportunity to create a new diagnostic framework for the disorder. Acute clinical observation allowed Osler to produce a remarkably coherent description of diabetes at the end of the 19th century.33 He recognised many of the aspects of the disease that we still use to define the disorder today, such as the elevation of sugar levels in the blood and the urine. He recognised also, however, the heterogeneity in the disease, its tendency to occasionally appear in families, and its occurrence in both young and elderly populations. Osler also had some inkling as to the major environmental factors that might contribute to the disease, alluding to both obesity and a sedentary lifestyle in his description of the disorder. However, he could make little further progress in defining this set of diseases as even the most rudimentary principles of the control of glucose were not recognised at that time.
The recognition of the role of pancreatic islets and insulin in the regulation of blood glucose provided a more fundamental understanding of the physiological processes that might be disturbed in this disease, but disease mechanisms still had not been clearly delineated and the role of the beta cell became overshadowed by the view that insulin resistance in the periphery accounted for the pathogenesis of this disease. In the 1970s, the characterisation of the genetic contribution by HLA alleles to the juvenile form of the disease (now type 1 diabetes) was described. This insight helped define a distinct subset of diabetes that involved autoimmune response, controlled by immune response genes in the HLA. This conclusion has been validated repeatedly since then and most of the more recently recognised genetic components of this autoimmune form of the disease contribute to the regulation of the immune response,12 affirming the power of genetics to identify mechanistically distinct subgroups in a heterogeneous disease.
Further progress in the characterisation of diabetes disease mechanisms came with a series of studies focused on Mendelian forms of diabetes, originally classified as maturity onset diabetes in the young (MODY).34 These studies have identified six loci associated with forms of MODY (GCK, HNF1A, HNF4A, HNF1B, IPF1, NeuroD1), often demonstrating distinct clinical features including differential response to therapy.34 Other rare forms of the disease such as neonatal diabetes have now also been genetically dissected into 12 genetic groups, all of which can now, to a large extent, be recognised as clinically distinct.34,35
With the advent of genome-wide association studies, a plethora of common variants have also now been associated with type 2 diabetes in populations.36–38 There appears to be no genetic overlap in type 1 and type 2 diabetes. To date,37 type 2 diabetes susceptibility loci have been identified, accounting for 10% of the heritability of this disease.
These GWAS data reveal important insights into disease mechanisms and pathophysiology. Physiological studies have produced contradictory data on the relative role of insulin resistance and obesity or beta cell dysfunction in this disease but, to date, the genetics strongly argue that genes affecting the beta cell are by far the clearest set of factors responsible for this disorder. Interestingly, only one gene identified to date is associated with abnormal insulin sensitivity associated with obesity and a handful of others are associated with reduced insulin sensitivity in non-obese populations.38,39 Many of the remaining genes are associated with beta cell dysfunction including cell cycle control of the beta cell and transcriptional regulation. Virtually all the results from studies of Mendelian forms of the disease and GWAS studies have described pathways previously unpredicted and uncharacterised in diabetes, demonstrating the very large gap in our understanding of disease mechanisms in this disorder. Together with the work on autoimmune forms of the disease, we now have a more complete understanding of the pathways that may be involved in the disorder, moving us one step closer to a more coherent classification system of this profoundly heterogeneous disorder.
The contributions of the genetics emerging from GWAS studies have played a major part in our understanding of disease biology and have opened up new opportunities for defining and treating disease. The contribution to heritability of the individual genetic variants emerging from GWAS studies is, however, small and together the variants have been shown to contribute only about 10% of the heritability in diseases such as type 2 diabetes and 20% in Crohn's disease. This is insufficient to contribute significantly to disease prediction and, in many cases, is less predictive than other known markers. How much the contribution to heritability is likely to rise as more common variants are detected and as existing associations are resolved to the functional variant responsible has been the subject of much discussion.40,41 Optimistic estimates suggest that up to half the heritability may be in the genome in this form, but whether or not it is accessible remains uncertain. There is also an important remaining question of whether our original estimates of heritability are valid or whether they have been overestimated due to shared environmental factors in twins. In any event, it seems likely that there remains heritability to be deciphered that will add further to our understanding of disease and its definition, but predictive testing using genetics is still a long way in the future.
The ‘black hole’ of genetic susceptibility
The premise that common genetic variants accounted for much of disease susceptibility underpinned the strategy to use common SNPs for GWAS studies. This proved to be a successful strategy but, to date, has provided incomplete understanding of heritability. The extent to which the heritability sits with common SNPs that are difficult to detect is currently the subject of some debate.40,41 Obvious additional places to look for the remaining heritability are less common genetic variants not detected by the SNP panels used for GWAS and even rare or ‘private’ mutations that might be found in single individuals or within a single family. Indeed, there is no reason to believe the genetic architecture of all diseases will be the same and, in some diseases such as autism, little evidence for common variants exists.42 While the common variant SNPs reflect polymorphisms selected because of a selective advantage in populations, rare variants may be recent variations and may be maintained by genetic drift. Such rare mutations, however, might also have significantly stronger penetrance in mediating disease pathophysiology. Until recently, the search for such rare variants was limited by the availability of high throughput sequencing technology. Nevertheless, there was evidence from conventional sequencing approaches that such rare variants existed and contributed significantly to individual variation. In 2004, studies conducted by Hobbs and her colleagues of genes known to cause low levels of HLA-C demonstrated that the frequency of non-synonymous sequence variants in a population with low levels of high-density lipoprotein cholesterol (HDL-C) were significantly more common than in the high HDL-C population (16% vs 2%) and were shown also to be biochemically important, and hence likely to have high penetrance.43
A second important line of evidence supporting a role for rare variants has come from studies of diabetes where a number of genes have been demonstrated to have highly penetrant rare variations that segregate in families, usually with MODY type diabetes. When GWAS studies have been performed for common variants, these have often appeared in many of the same genes.38 The definition of ‘rare’ and ‘common’ variants is, of course, arbitrary as a mutation uncommonly found in white MODY patients is recognised as a common variant in Oji-Cree Indian population.44 Five examples have emerged of common variants and rare variants in the same genes (PPARG, HNF1α, KCNJII, WF51 and HNF1β) in diabetes. These data suggest that rare and common variants may often populate the same genetic loci or pathways to mediate disease pathogenesis.
It is also clear that the genetic architecture of all common diseases need not be the same and some disease may be dominated by rare variants. Multiple rare variants in BRCA1 and 2, P53, PTEN, CHEK2, PALB2, ATM and BRIP1 all contribute to a risk of breast cancer, often as high-risk alleles.45 Similarly, hearing loss is commonly the result of rare alleles in multiple different genes and pathways rather than common variants.46 In addition to SNPs, structural mutations can contribute rare variants that mediate disease in certain patients. Neuropsychiatric diseases such as autism47,48,42 and schizophrenia49–51 demonstrate higher than expected numbers of rare copy number variants, often in pathways associated with relevant aspects of brain development, and GWAS studies have often failed to replicate in these diseases.42 There is much evidence to suggest that rare variants may add considerably to the heritability in some diseases.
The contribution of rare variants has on occasion been used as an argument against the utility of the search for common variants.45 The achievements of GWAS have, regardless of their absolute contributions to heritability, made a major contribution to the understanding of common disease. Most are robust and wholly reproducible and they have identified genes and pathways where we know rare variants are likely also to reside.38 It may prove that rare variants add understanding of heritability, particularly in some diseases. In other diseases, however, they may add less to the wealth of information already gleaned from common variations, particularly if they continue to be found in the same genes and pathways as those already identified by GWAS analysis.
The key technological development that allows rare variants and resequencing to be tractable are the new sequencing tools. These ‘next generation’ sequencing platforms are already in widespread use and the cost of sequencing a genome is falling dramatically, based on modifications and enhancements of these tools. A further generation of sequencing platforms is on the horizon, some of which do not require the use of strand synthesis to generate a signal, reducing cost and enhancing efficacy. Pore-based and electrochemical chip based strategies hold out the prospect that the price of sequencing will fall below the $1,000 per genome level.52,53 The challenge will be not in the generation of data, but in establishing which rare variants are functionally relevant and make a significant contribution to disease. Any single rare variant may require biochemical proof of involvement in disease pathogenesis, although the power of statistics may help with clusters of variants if a single variant has made a significant contribution to disease.
It may also prove that in terms of disease definition, prediction and monitoring, it will be more useful to use measurement of the underlying biological processes rather than simply genetics. Pathways and their biological outcomes must be altered by multiple genetic variants, both rare and common, as well as the response to environmental exposure. Where possible, therefore, the best approach to predicting, defining or monitoring disease may be to use biology as an integrated measure of the genetic factors. For example, assessment of Th17 populations will reflect the wide range of genetic variation in the IL-23 pathway and may prove to be the most important marker of disease in disorders such as AS or CD. What is different now is that, often for the first time, we know where to look in biological systems for relevant causal effects.
Cancer, the exemplar for genomic medicine
Of all the common disease areas where genetics and genomics have demonstrated their utility, cancer has increasingly become the outstanding example. Although the disease shares a set of recognised biological features, cancer has long suffered from the lack of diagnostic precision that has limited the accuracy of prognosis or response to therapy. Cell phenotype does not reflect the range of molecular events driving a tumour, nor does a diagnostic framework based on organ involvement properly reflect events shared between tumours at different sites. It may prove that the organ-specific basis of diagnosis may eventually be superseded by an understanding of the disrupted pathways. For example, the common mechanism of BRCA1 and 2 cancer in breast and ovarian cancer has already proved more important for prediction and possible therapy than the organ involved.54
From a molecular perspective, there are important differentiating features of cancer genetics that make it more tractable for discovery scientists. While most diseases, including cancer, have their origins in germ line genetic variation combined with environmental factors, cancer is associated with a further set of genetic variations – those found somatically within tumours. The drive behind disease progression and other clinical features of the disease, including metastasis and metabolic disruption, are often mediated by these genetic variants in tumour that are in part from the genome instability that accompanies the disease. Cancer, after it is initiated, becomes largely a genetic disease.
All the tools that have been used to discover genetic variation and define disease more precisely over the past 20 years have been used successfully in cancer. As in other disorders, genetic linkage studies and positional cloning were successful in identifying a range of highly penetrant germ line variants associated with susceptibility to familial cancers. Prominent among these were the breast cancer variants at BRCA1 and BRCA2.55,56 These and other uncommon forms of familial cancer proved tractable by classical molecular genetic approaches and have already reached the clinic in terms of screening individuals with strong family histories. They have also helped to define disease entities which are both mechanistically distinct and tractable with specific forms of targeted therapy. PARP inhibitors for BRCA1 mediated disease, for example, target the DNA repair pathway active in BRCA1 tumours by inhibiting poly (ADP-ribose) polymerase.57 In this example and in many others, such as EGFR, B-RAF, PTEN and PI3 kinase, the presence of genetic variants defines both drug targets and patient subpopulations for therapy.
As with other common diseases, there has also been an explosion in the discovery of common variants associated with a wide range of different cancers using GWAS studies. Again, these germ line effects are highly reproducible across large samples and have increased our understanding of aetiologic pathways. The variants provided by GWAS studies again provide little predictive power to identify individuals at high risk of cancer. The seven original breast cancer variants,58 when studied in large populations, provide a combined risk of breast cancer of two-fold, roughly equivalent to the risk associated with having one affected first-degree relative and considerably less than that associated with having two affected first-degree relatives with the disease. Nevertheless, these studies have provided considerable insight into the mechanism associated with the development of cancer.
It is, however, the characterisation of somatic variants in cancer that has provided some of the most clinically relevant information, identifying with precision drug responsiveness and natural history. These insights have emerged as sequencing technology has been applied to tumour samples. Sequencing exomes of tumours for variants has revealed a host of new important genetic associations. To date, more than 100 tumours have been sequenced revealing on average 40 mutants per cancer.59 With rapidly developing malignancies, such as acute leukaemia, fewer mutations are seen than in those tumours that have evolved over many years (colorectal cancer or pancreatic cancer). It is likely that many of the mutations are carrier mutations related to genomic instability and that only a few variations are responsible for driving tumour behaviour. Vogelstein has proposed, on the basis of the available genome sequences, 327 tumour suppressor or oncogenes in malignant disease that can be implicated in the generation or behaviour of these tumours.59 This set of mutated genes all relate to biological pathways suspected of being involved in tumorigenesis.60 Other genetic variants may also emerge that fall outside our current understanding of tumour biology, but it is increasingly clear that it will be possible, in many tumours, to define the major pathways that have been disrupted or altered by genetic alterations. Eventually, whole genome or exomic sequencing of tumours is also likely to provide insights into secondary behaviour of tumours including the ability of a tumour to metastasise to specific organs.
The genetic characterisation of gliomas has clearly demonstrated an important role for genetics in characterising specific subtypes of organ-specific cancers. Gliomas have been recognised for some time to have alterations in genes such as TP53, PTEN, DKN2A, and EGFR. Using genome-wide exomic sequencing, mutations have also recently been identified in the isocitrate dehydrogenase 1 (IDH1) gene in 12% of glioblastomas.61 Interestingly, these mutations affect only a single codon (codon 132) of the IDH1 gene and have also been recognised at position 172 in the IDH2 gene.62 Functionally, these two residues both sit in the active site of their respective enzymes and create hydrogen bonds with the substrate. We now know that these genetic variants produce a neoenzyme acting to produce excessive amounts of 2-hydroxygutarate in the neuronal cells,63–65 itself recognised to be associated with malignancy. The high frequency of this mutation and its presence, particularly in secondary glioblastomas, creates a new mechanistic paradigm for the conversion of primary gliomas to secondary tumours. The presence of this variant is mechanistically important for disease pathogenesis and is also associated with a different natural history amongst glioblastoma patients.62 The potential for novel therapies targeted at the neoenzyme is clear. This example of a role for cancer metabolism in pathogenesis reflects again the principles of chemical individuality as described in metabolic disease by Garrod. IDH mutations are another perfect example – this time of tumour individuality.
Cancer has become the paradigm for a more personalised approach to medicine.66 Because of our abilities to stratify this disorder mechanistically at a molecular level, it has also been possible to identify individuals who are likely to respond to particular forms of therapeutic interventions. There have been several examples of this approach to patient stratification over the past ten years. The development of imantinib, the kinase inhibitor specific for BCR ABL which produces dramatic responses in individuals with chronic myeloid leukaemia, is a targeted therapy specific for individuals with the cytogenetic abnormality that leads to constitutional activation of this kinase.67 This genetically modified target is both the drug target and the tool for defining responsive patients. The development of trastuzumab has also benefited from the ability to identify patient populations in which there is expression of Her2 on the surface of breast cancer cells.68 This effect, mediated by duplication of the Her2 gene, can now be detected using cytogenetic tools and again provides both the drug target and the mechanism for defining the response population. The improvement in efficacy from such agents in this personalised medicine approach has demonstrated the likely direction of cancer therapeutics in future years, from non-specific cellular inhibitors to those that are much more specifically directed. More recently, the recognition that epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors are only efficacious in the presence of wild type Kras69 has greatly enhanced their therapeutic efficacy and demonstrated the importance of understanding the role of individual mutations in cancer signalling pathways. Mutations in EGFR itself also dictate response to some EGFR inhibitors,70,71 demonstrating how one of the earliest EGFR antagonists, IRESSA (Gefitinib), failed in trials due to lack of efficacy but, in genetically stratified patients with lung cancer, is a highly effective therapy.72,73 A new class of compounds specific for the kinase Braf has also been developed and demonstrates striking therapeutic efficacy in patient populations. 40% of patients with malignant melanomas have a mutation in Braf (V600E) that is responsible for its malignant characteristics.74 A host of Braf inhibitors have been developed targeting this by signalling factor. One such molecule which targets only the mutant Braf V600E has demonstrated impressive results in early trials (Fig 2).75 Similarly, a new agent targeting the fusion that results from an inversion on chromosome 2 of EML2 and ALK have shown early promise in non small cell lung cancer where the fusion is present in approximately 5% of patients.76 Genetic analysis of tumours is providing remarkable insights into the pathways responsible for tumour pathogenesis and growth and, in turn, is creating the opportunity to precisely target components of these pathways with new medicines. Personalised medicine of this kind is likely to become widespread as the efficacy and safety delivered from such targeted medicine is likely to be required of most new medicines.
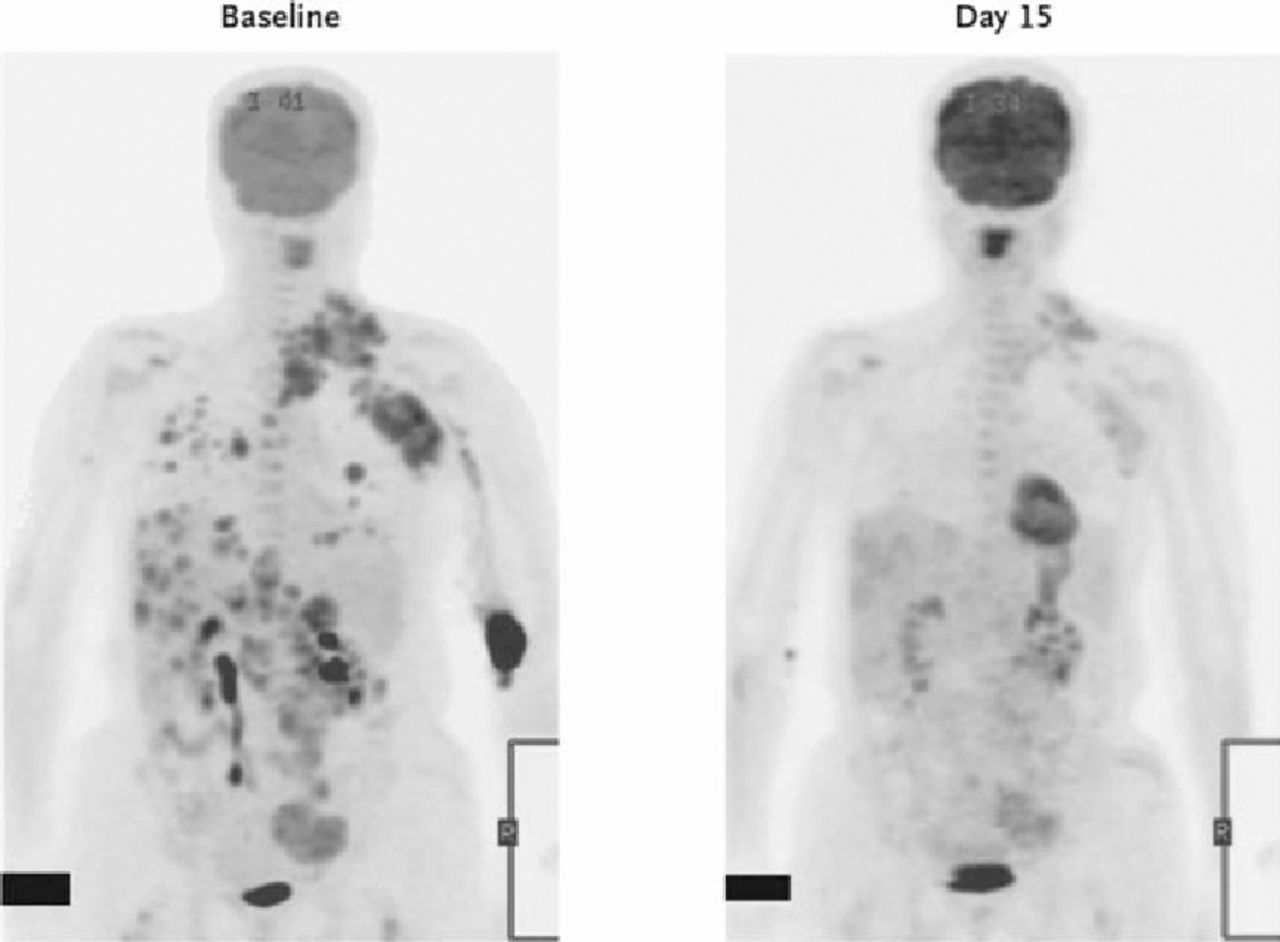
Fig 5. Efficacy of PLX4032 at Braf inhibitor demonstrated by positron emission tomography of 18F-fluorodeoxyglucose (FDG) at baseline (left) and on day 15 (right) of treatment. Reproduced with permission from Flaherty KT, Puzanov I, Kim KB et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363:809–19. Copyright © 2010 Massachusetts Medical Society. All right sreserved.
Genetic tools supplied to cancer are providing extensive insights into the pathways involved in pathogenesis. These observations will provide a better understanding of tumour natural history and, importantly, will also provide opportunities to target new therapies at individual patient subpopulations. This rapid progress is already creating a new diagnostic framework for cancer that is creating widespread redefinition of the disease.
Back to the environment
In a few relatively uncommon conditions, genetics should be sufficient to provide a complete understanding of the pathophysiology of the disease. Most conditions, however, will rely on an understanding of the genetics and biochemical pathways responsible for determining susceptibility and also an understanding of the environmental exposures that, in most common complex diseases, contribute at least 50% to the risk of disease as assessed in twin studies. The search for additional determinants of heritability will continue exploring further common variants as well as rare variants around the genome, while the search for environmental factors in disease has, with only a few exceptions, just begun.
Environmental factors can occasionally act directly on DNA to produce their effects, as in the case of external mutagens or through epigenetic modification of DNA. More commonly, however, it is likely that the environment interacts with the biochemical pathways associated with disease pathogenesis that are also influenced by genetic variation. As with genetic studies, associations between individual environmental factors in disease can be established at a statistical level by comparing those with disease to those who are unaffected but, ultimately, an understanding of how these factors interact with biological processes will be essential for a more complete understanding of how disease aetiology occurs.
It is likely that some of the challenges that have prevented environmental studies from identifying more than a handful of powerful environmental effects contributing to disease can be attributed to the heterogeneity in disease populations where more than one biological pathway may account for a particular disease phenotype. Characterising environmental exposures, therefore, in the context of disease subgroups based on genetic variants in specific pathways, may provide an opportunity for more powerful studies to emerge. Defining these patient subgroups based purely on genetic variation may prove to be difficult, however, as multiple different genetic determinants can contribute to individual biological pathways and it is the integrated impact of these genetic variants on those pathways that is likely to determine whether it is contributing significantly to disease biology. It may prove important, therefore, to analyse environmental exposure data with an understanding of the level of activation of whole pathways rather than individual genetic variants. For example, it may be important to understand which environmental factors contribute to Crohn's disease or ankylosing spondylitis in the context of elevated TH17 cell levels27,28 that might arise from a range of genetic variation in the IL23 receptor signalling pathway.
The area where our best understanding of environmental factors and their contribution to disease has emerged has been in the field of infectious disease. Infectious pathogens are now recognised to be responsible for a host of clinical syndromes. Clinical phenotypes are in part determined by the genetic make-up of the host, as individuals can show widely varying responses to individual pathogen exposure. The role of infectious pathogens in other diseases such as cancer is also now well recognised and is in part determined by host responses. The importance of infections in selecting much of the genetic variation that is associated with autoimmune disease is also well-recognised. Although the role of viruses has often been proposed as an initiating factor in autoimmunity while never formally proven, suggestive associations between alleles of genes that encode viral receptors such as the IFIH1 gene and type 1 diabetes or SLE, continue to indicate a potential role of pathogens in the mediation of some of these disorders. The interactions between environment (in this case, pathogens) and genetics involves selection of host and pathogen genes that also on occasion produce pathological outcomes.
It is clear that the availability of genetic markers that define pathways associated with disease may improve our ability to track and characterise some of the environmental factors that also contribute to pathogenesis. There are a variety of strategies that are likely to be used for this purpose, some of which are already yielding valuable results. Historically, the approach to characterising the environmental factors in disease has been to undertake large population studies and to characterise environmental exposures in individuals who develop disease and compare these to those who do not. Although such approaches have identified powerful environmental factors with large effects, such as smoking and asbestos exposure, these approaches could be considerably enhanced once the disease heterogeneity has been reduced by distinguishing patient subpopulations. The prospect, therefore, of using both genetics and environment to characterise genetic and environmental risk factors in disease and, ultimately, to identify gene–environment interactions, has resulted in the creation of large prospective cohorts with appropriate biological material collected to allow genetic analysis as well as more conventional environmental exposure information. Studies such as UK BioBank77 and the Kadoori study will provide large populations where environmental exposures have been collected prospectively to reduce the risk of ‘reverse causality’ and to allow the systematic analysis of genetic determinants alongside the environment. This remains a potentially exciting opportunity for the future. Given that genetic variants are likely to produce integrated signatures on disease pathways, it will also be important to characterise patient populations in whom the activity of biological pathways can be analysed systematically, both in normal patient populations and those with disease. These studies are now increasingly being performed in more highly characterised populations of patients who have been genotypically selected for the analysis of their pathways (eg Cambridge Bio-resource and Oxford Biobank).78,79 Well-characterised patient populations become a crucial component of these studies that will provide information about biological markers of perturbed biological pathways that can subsequently be used for characterisation of large patient cohorts in search of environmental factors of disease.
One further approach to understanding the role of environment in interacting with disease pathways is the use of animal models which have been cleverly constructed to incorporate disturbances of genetic pathways known to predispose to disease in man. The utility of animal models as predictors of human disease has been limited, in part because the relevant pathways and components that contributed to human disease pathogenesis have not previously been available to those constructing appropriate animal models. Now that some of those determinants and pathways have been defined, it becomes possible to develop and utilise much more appropriate animal models. One powerful advantage of these models is that the interaction of environmental agents and biological pathways can be tested in ways that are not possible in man. The detailed analysis of genetic determinants of autoimmune disease encoded within the HLA have best been analysed in the context of mouse models where individual components of linkage disequilibrium blocks can be studied in isolation or in combination. This has revealed significant evidence of epistasis between HLA alleles at different loci that could not be dissected because of the presence of near complete linkage disequilibrium in human populations.80 Similarly, murine models of disease have now been created for other inflammatory conditions such as Crohn's disease.23 For example, when hypomorphic variants of the ATG16L1 autophagy gene have been created in mice, these animals display interesting pathological changes in Paneth cells, in their gastrointestinal tract with evidence of abnormal packaging of antimicrobial granules and excessive transcription of pro-inflammatory cytokines. These changes are consistent with the picture seen in patients possessing the ATG16L1 variants but not controls. This model has recently been extended to demonstrate the role of a murine Norovirus in producing these Paneth cell abnormalities. These mice show a pathological response to dextran sodium sulphate that requires the presence of cytokines TNFα and interferon gamma. Treatment with TNFα blocking antibodies dramatically reduced the features of this syndrome, consistent with the utility of these agents in human Crohn's disease. The model also demonstrates an important role for commensal bacteria as broad spectrum antibiotics can considerably reduce the clinical syndrome. Such models provide powerful evidence that multiple environmental factors may interact with particular genetic variants to create the conditions under which a disease can occur. They also provide evidence that environmental factors may be multiple and complex, indicating why so few have emerged to date.
Conclusion
For most of the history of medicine, little or no effort has been put into characterising the scientific basis of disease. When the opportunity arose to link infectious pathogens with disease pathology and clinical syndromes, it led to a period of remarkable enlightenment in clinical practice so well described by Osler in The principles and practice of medicine. Shortly thereafter, Garrod laid out for the first time the underpinning principles by which we now recognise disease to occur. The combination of genetic factors and environmental exposures provides the crucial mix to produce the biological events that lead to individual diseases and diseases in individuals. The opportunity to build on this intellectual framework of disease laid down by Garrod did not, however, emerge until the end of the 20th century when, for the first time, genetic tools became available to characterise the pathways and processes involved in a wide range of common complex diseases. We now know where the process of dissecting genetic factors is likely to end, with a greatly improved understanding of the determinants of heritability of disease and the rationale behind why some individuals suffer from particular ailments while others do not. Crucially, these genetic studies have already provided remarkable insights into the mechanisms and processes responsible for many disease pathologies. These insights are, for the most part, novel, indicating how far we as a profession have been from truly understanding the fundamental scientific principles of the disorders we treat. We now have an understanding of what some of these mechanisms are likely to be, providing an opportunity to explore and develop the fundamental events in disease pathogenesis as opposed to clinical features and phenotypes.
One of the most exciting outcomes of these studies is that it creates a framework of linking causality with disease definition and moves away from a disease taxonomy which has been unrelated to the events associated with pathogenesis. This is an important step for all of medicine as it will underpin all aspects of clinical practice with scientific principles and the understanding of disease pathways. All aspects of natural history will become clearer, from disease initiation to late stage progression. It will also provide enormous opportunities for the development of therapies targeted at appropriately defined patient populations with specifically characterised diseases creating an era of more personalised medicine. Finally, it will provide the platform upon which to search for the key environmental factors that interact with the proteins and biological pathways that emanate from genetic factors to produce disease. This potentially holds additional opportunities, both for more refined disease definition and also for preventative medicine and public health. There have been few occasions in the history of medicine where such opportunities exist to totally transform both our understanding of disease and the way we practise medicine. The potential for scientific discovery and the health of patients with virtually every form of disease could now be realised as we continue this systematic effort to redefine disease.
Footnotes
The Harveian Oration is delivered annually at the Royal College of Physicians under an indenture of William Harvey in 1656. This article is based on the 2010 oration delivered on 19 October 2010 by Sir John Bell, president of the Academy of Medical Sciences
- Royal College of Physicians








{kind=link}
{kind=link}