
Lesson
A 47-year-old bus driver was admitted following an out-of-hospital cardiac arrest. Paramedics delivered direct current cardioversion (DCCV) for ventricular fibrillation (VF) and spontaneous circulation was restored. The past medical history was of recently diagnosed type II diabetes. His elder brother was diagnosed eight years previously, at the age of 46, with MD. This was genetically confirmed by a cytosine-thymine-guanine (CTG) trinucleotide repeat expansion at the dystrophia myotonica protein kinase (DMPK) gene locus. The brother had a history of first degree heart block and previously had been labelled as ‘opiate toxic’ after receiving a general anaesthetic with muscle relaxant. Their father had died aged 65 from pneumonia, and was described to have had frontal balding and muscle weakness. The father had a diagnostic label of ‘multiple sclerosis’ (MS). Neither paternal grandparent was known to have muscle weakness and the maternal family history was unremarkable.
In the emergency department, recurrent episodes of non-sustained ventricular tachycardia (NSVT) were terminated by intravenous amiodarone. The electrocardiogram (ECG) showed left bundle branch block (LBBB) and a QTc interval of 509 milliseconds (Fig 1). His Glasgow Coma Scale (GCS) remained low at 7/15, requiring intubation and ventilation. An urgent computed tomography (CT) brain scan was unremarkable.
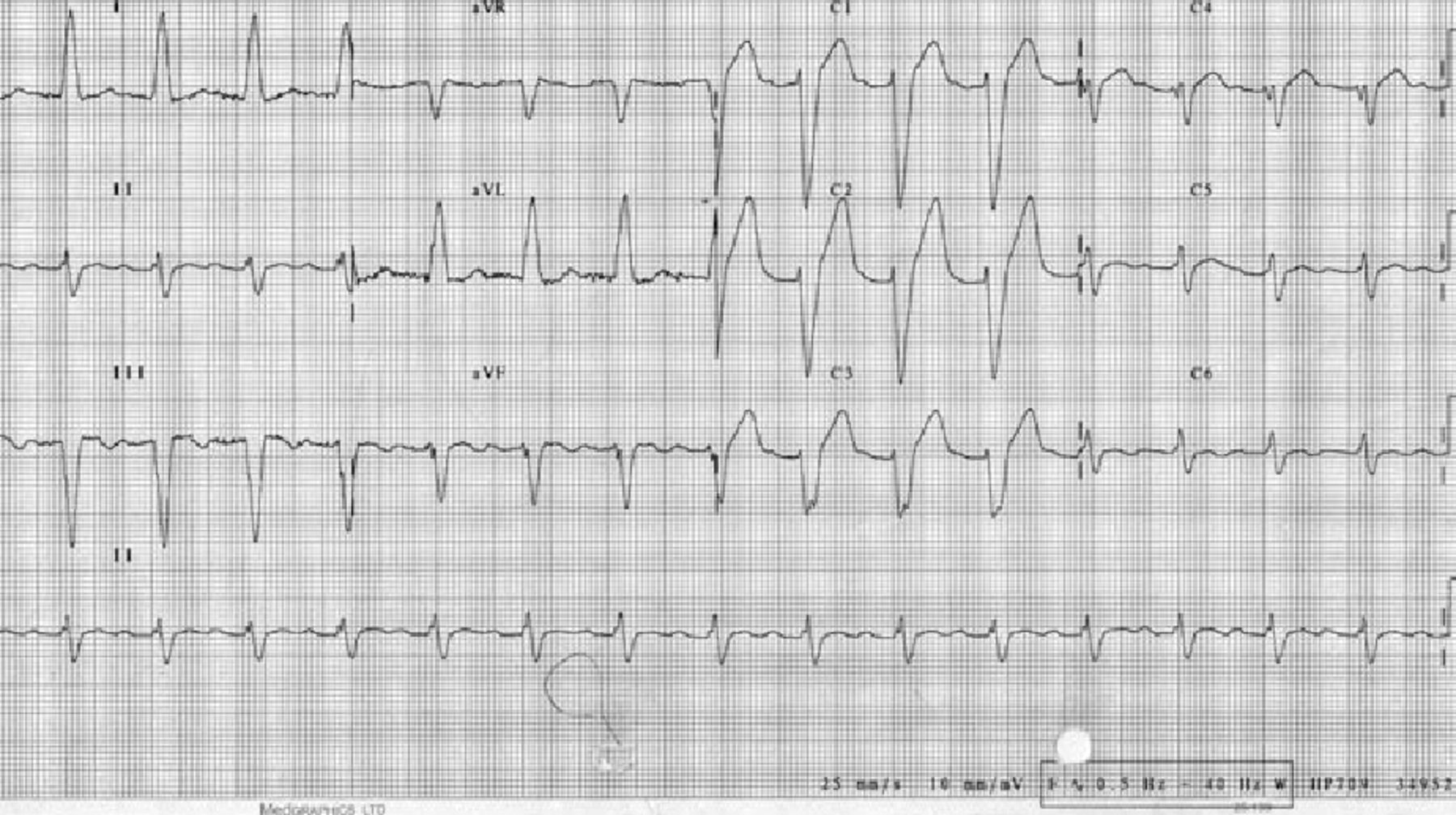
Resting 12-lead electrocardiogram showing left bundle branch block and a QTc of 509 milliseconds.
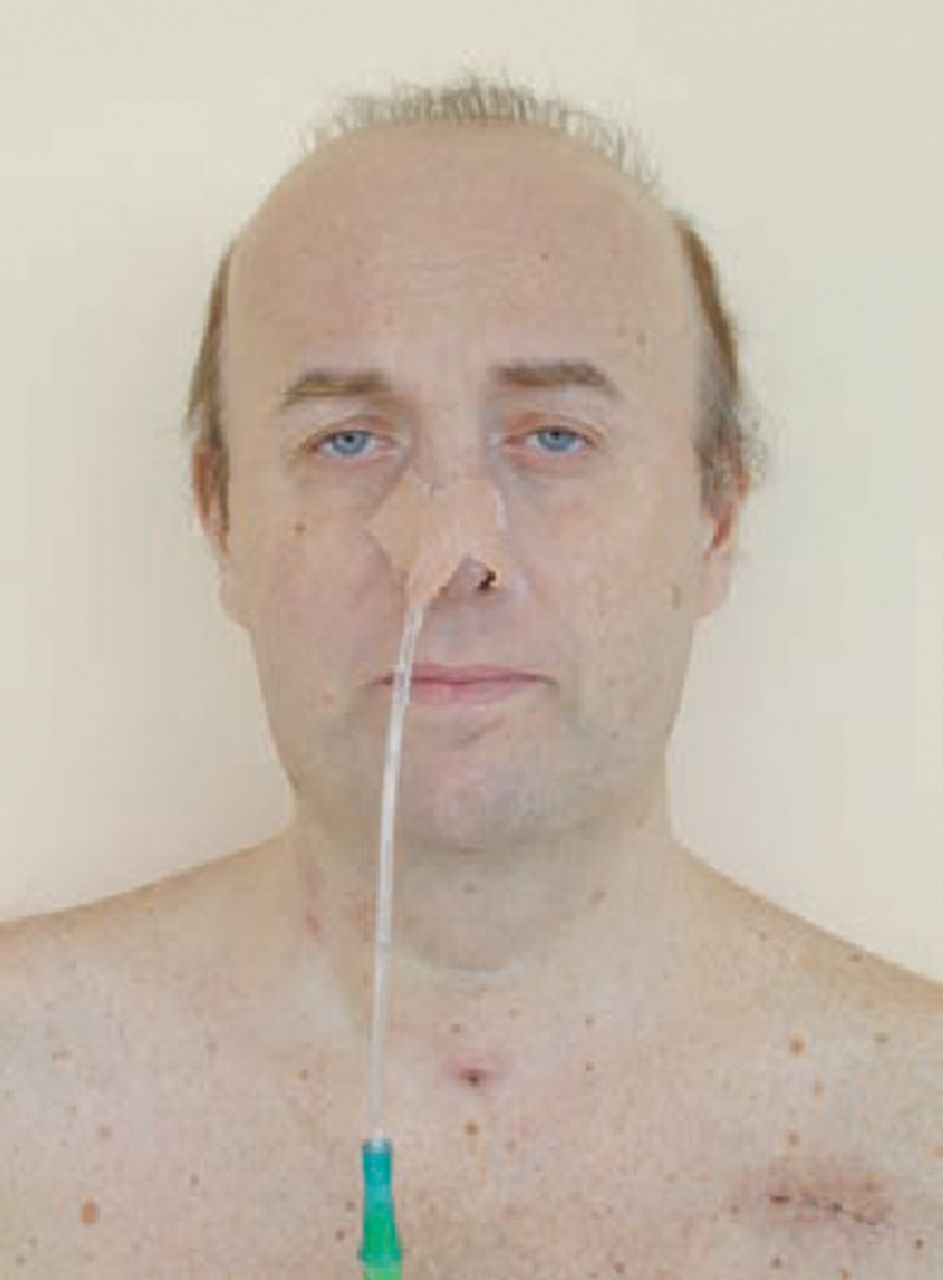
Admission to the intensive care unit was prolonged and complicated by difficulty in ventilator weaning which necessitated a tracheostomy. Post-extubation, marked dysphagia was noted and videofluoroscopy showed reduced cricopharyngeal opening, severe saliva pooling and evidence of on-going aspiration. Nutrition was therefore provided via nasogastric tube. Coronary angiography showed normal coronary arteries. A repeat CT brain scan, to exclude brain injury contributing to his protracted recovery, was again normal. Clinical examination suggested myotonic dystrophy (Fig 2). He had a positive grip test, percussion myotonia and his speech was slurred. The diagnosis was confirmed by a neurologist.

Photograph of patient with features of myotonic dystrophy: frontal balding, bilateral ptosis, wasted temporalis and sternocleidomastoid muscles. There is a nasogastric tube in situ to reduce risk of aspiration due to abnormalities in swallowing. A scar is visible from recent automatic implantable cardioverter-defibrillator implantation following cardiac arrest. A scar from the tracheostomy is also visible.
An automated implantable cardioverter defibrillator (AICD) was inserted for secondary prophylaxis against VF. A transthoracic echocardiogram showed left ventricular dimensions at the upper limits of normal, moderate systolic function (ejection fraction of 55%) but marked dyssynchrony.
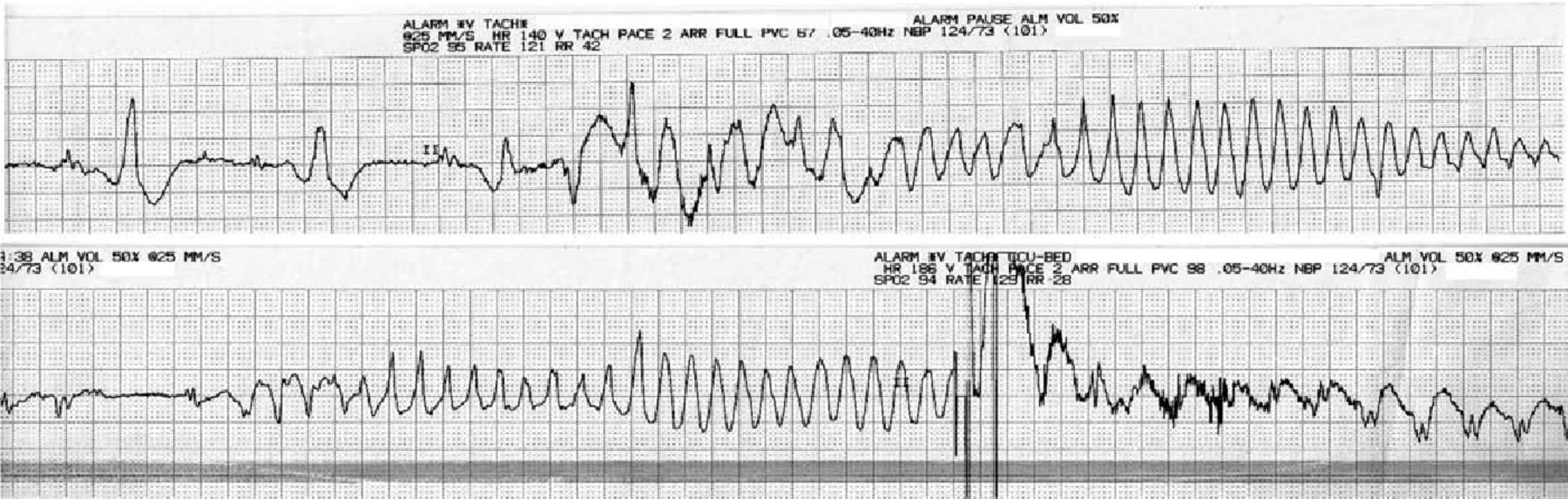
Recovery involved regular dysphagia exercises on a neurorehabilitation ward. An aspiration pneumonia associated with Enterobacter aerogenes bacteraemia complicated recovery and was treated with clarithromycin and co-amoxiclav. This was followed by episodes of polymorphic VT (‘Torsades de Pointes’ Fig 3) resulting in AICD discharge. This was stabilised with beta-blockade (bisoprolol) and an ace-inhibitor (ramipril) was added to optimise cardiac function. The patient is likely to require placement of a percutaneous enterogastrostomy (PEG) tube because an ongoing risk of aspiration remains.
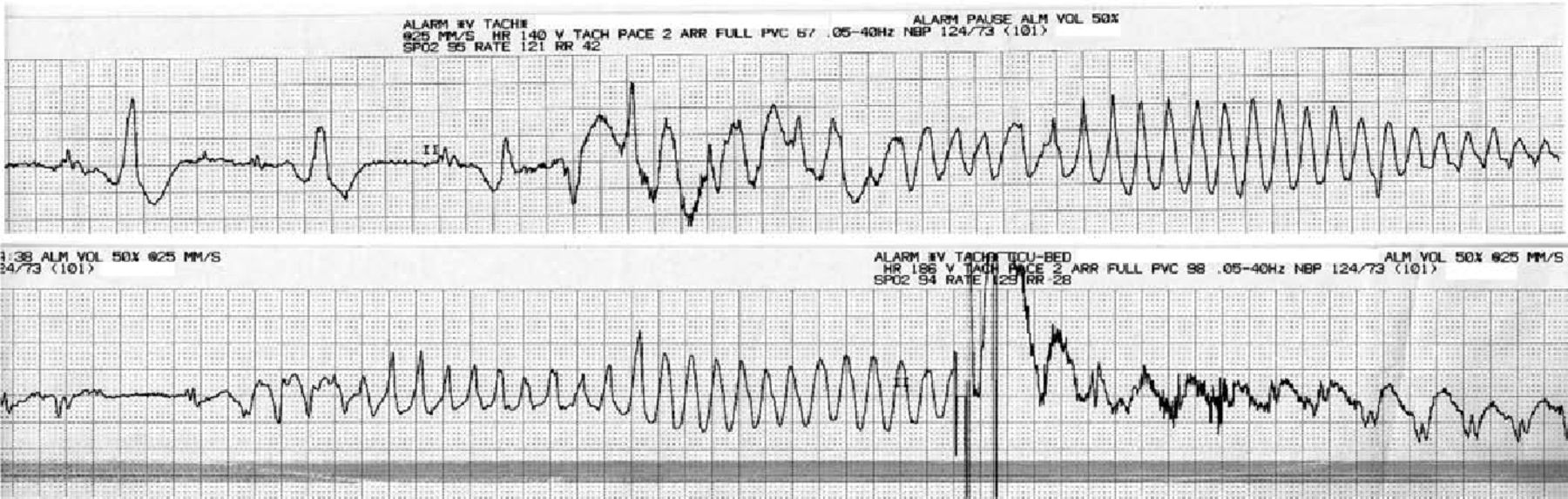
Electrocardiogram rhythm strips showing recurrent polymorphic ventricular tachycardia (‘Torsades de Pointes’). On the lower strip, a shock was delivered by the automatic implantable cardioverter-defibrillator.
Discussion
The genetic defect in MD is a trinucleotide repeat expansion at two different loci representing two clinical variants. The MD1 form accounts for 98% of cases and is due to a CTG trinucleotide repeat at the DMPK gene on the long arm of chromosome 19. The DMPK gene encodes for a myotonin protein kinase involved in cell signalling and plays structural roles in skeletal and smooth muscles.2 Successive generations have an expansion in the number of trinucleotide repeat, known as anticipation.2 This clinically manifests as offspring presenting with more severe forms of the disease at an earlier age. A childhood onset variant is known as congenital myotonic dystrophy (CMD). The MD2 variant is due to trinucleotide repeats on chromosome 3, at the CNBP gene (‘CCHC-type zinc finger, nucleic acid binding protein’). Disease severity and progression tends to be greater in MD1. The patient presented almost certainly has MD1, given the genetic confirmation within the older sibling. The father must also have had MD which was misdiagnosed as MS over 25 years ago.
MD affects cardiac muscle and smooth muscles of the gastrointestinal tract resulting in significant morbidity and mortality. The main cardiac complications are conduction defects, caused by myocardial fibrosis of the conduction pathways.3–5 The degree of fibrosis at autopsy correlates with the degree of ante-mortem ECG abnormalities.5 The surface ECG can show prolonged PR and QRS durations.6 Arrhythmias are common, including sinus node dysfunction, atrial tachycardias, atrial fibrillation and flutter.5,6 There may be progression to heart block and life-threatening ventricular dysrhythmias or asystole.5,6 Arrhythmogenic sudden cardiac death (SCD) has attributed up to 29% of sudden unexpected mortality in MD but this may be underestimated due to difficulties in post-mortem diagnosis.8 Conduction defects are reported as being more frequent in the MD1 than the MD2 form.9 A prolonged QTc with inherent repolarisation abnormalities is a recognised mechanism for the underlying ventricular dysrhythmias and spontaneous polymorphic VT is associated with MD.4 In this case the use of amiodarone and clarithromycin, both recognised as prolonging the QT interval, may have precipitated the polymorphic VT.
Myocardial fibrosis appears to progress at an unpredictable rate.3 Therefore, screening for arrhythmias and identifying those most likely to benefit from a pacemaker or AICD, can be challenging. There may also be familial propensity to SCD. Patients with evidence of bradyarrhythmias can be protected by simple pacemakers, however, SCD may still occur.7 AICDs can help abort life-threatening ventricular dysrhythmias; they are indicated for secondary prophylaxis and have been used for primary prophylaxis in high risk groups.6 However, their effectiveness in reducing sudden death in MD remains unclear, and the importance of respiratory failure due to respiratory muscle weakness as an alternative cause of death must be emphasised.
The 12-lead ECG is a useful non-invasive method of predicting the risk of SCD in MD. An atrial tachycardia, PR interval greater than 240 msec, QRS duration greater than 120 msec, and second or third degree AV block are independent risk factors for SCD.6 An observational study of 406 patients with genetically confirmed MD over a mean period of 5.7 years had 81 recorded deaths, of which 27 were sudden.6 Seventeen of 27 had post-collapse ECG recordings, of which nine (11% of total deaths) showed a ventricular tachyarrhythmia.
Transthoracic echocardiography often shows normal left ventricular (LV) dimensions, but reduced systolic function in both variants of MD; this is more frequent in MD2.9 Even when LV impairment is mild, there is often marked cardiac dyssynchrony.10 Both electrical (broad QRS) and myocardial factors for dyssynchrony are implicated.10 Dyssynchrony is associated with reduced exercise tolerance and a poorer prognosis. If detected early, cardiac resynchronisation therapy can potentially improve survival and reduce heart failure symptoms, as in conventional heart failure therapy.10 Global cardiomyopathy remains unusual in MD.3
MD also affects the smooth muscles of the GIT causing dysphagia due to oesophageal dysmotility.11 Abdominal cramps, constipation and sometimes diarrhoea result from small and large bowel dysmotility. In severe cases, megacolon or an ileus may develop.11 CT scans or ultrasound scans of the abdomen often reveal no structural cause for these abnormalities. The extent of dysphagia and safety of swallow mechanism may be evaluated by a video fluoroscopy and/or modified barium swallow. In high risk patients, PEG feeding may be required to avoid aspiration.
Involvement of the diaphragm and other respiratory muscles is common and generally parallels the development of limb weakness.12 Supine dyspnoea and nocturnal hypoventilation can occur, while aspiration pneumonia can trigger respiratory failure. Abnormalities in central respiratory drive may also occur.12 MD patients are sensitive to hypnotics and paralysing agents used in anaesthesia, potentially prolonging ventilator dependency and this was demonstrated in this case. Acute respiratory failure accounts for the majority (30–75%) of MD deaths.8
This case illustrates the many myriad features of MD and illustrates the significant morbidity from cardiac and gastrointestinal dysfunction. It is suggested that family screening should be obligatory and that regular Holter surveillance should be performed. Particular vigilance is required with therapies that extend the QT interval given a propensity to polymorphic VT.
- Royal College of Physicians








{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.