
Key points
Vasculitis is defined as inflammation within blood vessel walls, with biopsy the diagnostic gold standard.
Vasculitis may be primary (autoimmune) or secondary to an identifiable underlying cause such as infection
Features common to the vasculitides include fever, night sweats, malaise, arthralgia, myalgia and weight loss. Additional features then vary according to the specific disease
The clinical information should be used to determine whether the vasculitis is primary or secondary
If the vasculitis is secondary, the underlying cause should be identified, with investigations targeted specifically at excluding infection, confirming the clinical diagnosis and assessing disease extent
The principle questions regarding treatment are: Does the vasculitis require urgent treatment? Is it secondary to an underlying treatable cause?
KEY WORDS: antineutrophil cytoplasmic antibody, biopsy, glomerulonephritis, primary, secondary
Patients with vasculitis may present for the first time to the medical take, therefore clinicians in acute medicine need to be familiar with the myriad of potential clinical features. This article covers the definition of vasculitis, its classification, clinical presentations, useful first-line investigations and briefly discusses initial therapeutic options.
Definition
Vasculitis is defined as inflammation within blood vessel walls, with biopsy the diagnostic gold standard. The clinical and pathological features are variable depending on the site and size of vessel affected. Vasculitis may be primary (autoimmune) or secondary to an identifiable underlying cause such as infection (see below). The aetiology and pathophysiology of the primary vasculitides are rarely known. The clinical and histological features often overlap and, to date, no classification systems in isolation has been satisfactory. The best known nomenclature of systemic vasculitides is based on the Chapel Hill Consensus Conference.1 More recently, the European League Against Rheumatism has suggested contemporary points to consider for the development of future definitions and criteria.2
Classification
Clinical
Primary systemic vasculitis
Clinical classification is based on vessel size, divided into large, medium and small vessel vasculitis:
large: temporal and Takayasu arteritis
medium: polyarteritis nodosa (PAN) and Kawasaki disease
small: Wegener's granulomatosis (WG), microscopic polyangiitis (MPA), Churg-Strauss syndrome (CSS), Henoch Schönlein purpura (HSP), cryoglobulinaemia and antiglomerular basement membrane (GBM) disease.
Secondary vasculitis
Secondary causes of vasculitis (frequent causes seen on the acute medical admission ward) include infection, drugs, connective tissue disease (CTD) and malignancy. Important causes of infection associated with vasculitis include subacute bacterial endocarditis (SBE) and meningococcal disease. Viral causes include cytomegalovirus, Epstein Barr virus, HIV, hepatitis B and C infection.3 Drug-associated vasculitis can occur with a wide variety of drugs, of which one of the best known is propylthiouracil-induced antineutrophilic cytoplasmic antibody (ANCA)-associated vasculitis.4 Another well recognised cause is hydralazine. Vasculitis has also been documented with biologic therapies (eg tumour necrosis factor-targeted therapies) following their increasing use.5
Vasculitis associated with CTD can occur, for example, in rheumatoid arthritis, systemic lupus erythematosus (SLE) and Sjögren's syndrome. Additional clinical features of these conditions may be evident. Type 1 cryoglobulins (monoclonal) can be seen as a manifestation of underlying haematological malignancy.
Histological
Histological classification is based on vessel size, distribution and type of inflammatory cell infiltrate so, by definition, requires a tissue diagnosis. Small vessel involvement is often non-specific, with lymphocytes, polymorphs and nuclear dust, described as leucocytoclastic vasculitis. Large vessel disease is predominantly granulomatous, as are some forms of small vessel vasculitis such as WG and CSS. Medium vessel vasculitis (eg PAN) is immune complex-mediated, as is vasculitis associated with SLE and cryoglobulinaemia. MPA is leucocytoclastic and HSP is associated with immunoglobulin (Ig) A deposition.
Immunological
Attempts to classify vasculitis according to the immunopathogensis, for example, ANCA-associated (small vessel) vasculitis, immune complex-mediated or granulomatous are summarised in Table 1 (Fig 1).
Immunological classification of small vessel vasculitis.

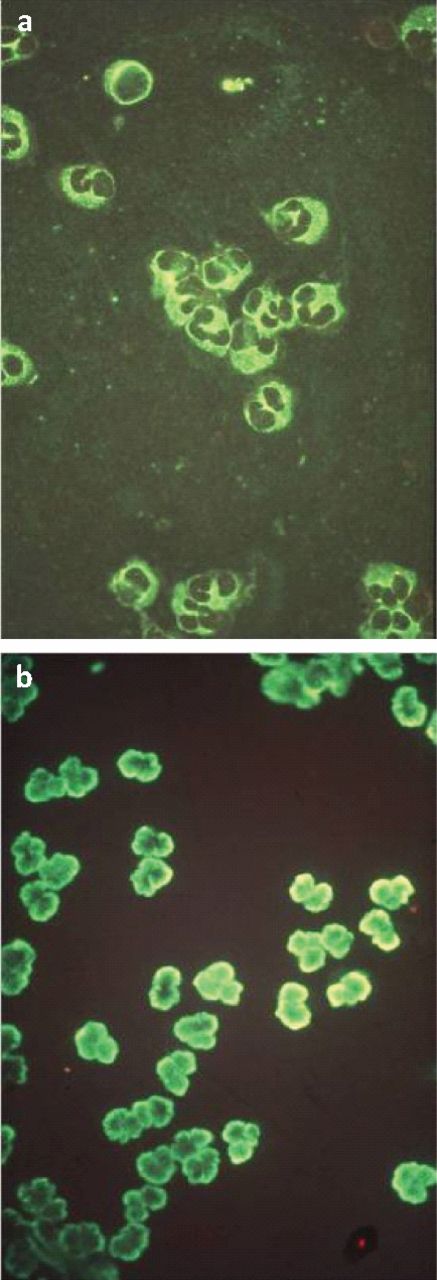
Antineutrophilic cytoplasmic antibody (ANCA) patterns by indirect immunofluorescence. (a) Cytoplasmic ANCA: positive staining in the neutrophil cytoplasm, with granularity and nuclear interlobar accentuation. (b) Perinuclear ANCA: positive staining accentuated around the neutrophil nucleus (ie perinuclear).
Clinical presentation
Features common to the vasculitides include fever (patients may present with pyrexia of unknown origin), night sweats, malaise, arthralgia, myalgia and weight loss - that is, systemic symptoms. Additional features then vary according to the specific disease:
Temporal arteritis is the most common form of primary systemic vasculitis with an incidence of 200 per million population per year.6,7 It tends to occur in patients over the age of 50, with symptoms including headache, facial pain, jaw claudication and, of most concern, the potential for sudden, painless, irreversible visual loss - hence the need for immediate treatment as soon as the diagnosis is suspected.
Takayasu arteritis is a much rarer large vessel vasculitis, predominantly affecting the aorta and main branches.8 It presents at a younger age, usually in females below the age of 40. Non-specific findings include erythema nodosum. More specific features include claudication and ischaemic symptoms, including cerebral ischaemia, loss of pulses, blood pressure discrepancy (>10 mmHg between arms), arterial bruits and aortic regurgitation.
PAN generally involves medium sized vessels and often presents with ischaemia or infarction of the affected organs. It predominantly affects the gut, heart, kidney and peripheral nerves. It is more common in males and can be associated with hepatitis B infection.6
Kawasaki disease, also known as mucocutaneous lymph node syndrome due to the pattern of clinical presentation, is typically a paediatric vasculitis and so unlikely to present in the adult medical take. The main concern is of coronary vessel involvement, with subsequent development of coronary artery aneurysms and cardiac ischaemia.
HSP is also more common in paediatric practice. Patients present with purpura of the lower limbs and buttocks, associated with haematuria, abdominal pain, bloody diarrhoea and arthralgia. Most cases resolve without progressive renal disease.
WG typically involves the respiratory and renal tracts, hence it can present in a number of ways to the medical team. Clinical symptoms can include upper respiratory tract infection, otitis media, tracheal stenosis, cough, dyspnoea and haemoptysis, including potentially life-threatening pulmonary haemorrhage. Renal disease occurs in approximately 80% of cases, with haematuria, proteinuria, hypertension and rapidly progressive glomerulonephritis. Other organ systems can also be affected including the eye and gastrointestinal (GI) tract.6
MPA predominantly presents with renal disease, with haematuria, hypertension and rapidly progressive glomerulonephritis. Pulmonary involvement includes pulmonary haemorrhage, but upper respiratory tract involvement is unusual. Rare manifestations include episcleritis and coronary disease.
CSS is associated with asthma and pulmonary infiltrates, eosinophilia, rash, mononeuritis, renal, cardiac and GI involvement. Asthma with eosinophilia is common, so there is a risk of overdiagnosis and careful attention should therefore be paid to identifying additional features. Conversely, CSS can potentially be unmasked in patients with presumed asthma when corticosteroid therapy is reduced following the introduction of other disease-modifying agents such as leukotriene receptor antagonists or omalizumab.9
Cryoglobulinaemic vasculitis typically presents with palpable purpura of the extremities, arthralgia and peripheral neuropathy.10 Glome-rulonephritis is common; GI, cardiac and central nervous system involvement is also reported. Cryoglobulins may occur in the context of lymphoproliferative disease, CTD and chronic infection such as hepatitis C. Cryoglobulins can be monoclonal, polyclonal with a monoclonal component or purely polyclonal depending on the underlying cause.
Anti-GBM disease typically presents with rapidly progressive glomerulonephritis and pulmonary haemorrhage,6 the latter especially in smokers.
Clinical history
Important factors to determine from the clinical history include symptom onset, course and duration, and preceding events such as infection and drug administration. Systems involved need to be established.
Clinical examination
Urinalysis is important in determining potential renal involvement. It is an easily overlooked component of the examination on the acute medical take.
Careful attention to cutaneous and nail fold changes may give clues: for example, splinter haemorrhages in SBE.
Joint examination is relevant when considering vasculitis/CTD.
Blood pressure may be elevated in relation to rapidly progressive glomerulonephritis. Significant upper limb blood pressure discrepancy may be found in large vessel vasculitis.
All pulses should be examined.
Cardiac murmurs may signify SBE or large vessel vasculitis and need to be taken seriously.
The presence of organomegaly may suggest underlying lymphoproliferative disease.
Peripheral neuropathy can be seen in small vessel vasculitis.
Fundoscopy should be undertaken to assess for retinal changes associated with SBE and hypertension.
Investigations
Basic
The clinical information should be used to narrow down the most appropriate investigations, determine if vasculitis is primary or secondary and, if secondary, then determine the underlying cause. Investigations (Table 2) are targeted at:
excluding infection
confirming the clinical diagnosis
assessing disease extent/organ involvement
supporting treatment options.
Additional, according to clinical presentation
These include:
Chest imaging to assess for cavitatory disease in WG, peripheral infiltrates in CSS, pulmonary infiltrates/haemorrhage in WG and anti-GBM disease. Bronchoscopy with bronchoalveolar lavage may be required (Fig 2).
Echocardiography to assess for vegetations in SBE, aortic regurgitation in Takayasu arteritis and large vessel vasculitis associated with CTD, and coronary aneuryms in Kawasaki disease.
Angiography to assess blood vessel anatomy more directly, looking for stenoses and/or aneurysm formation (eg in Takayasu arteritis and PAN) (Fig 3).
Biopsy. Tissue diagnosis can be confirmatory for vasculitis and may help guide treatment (eg temporal artery, renal, sural nerve or skin biopsy).
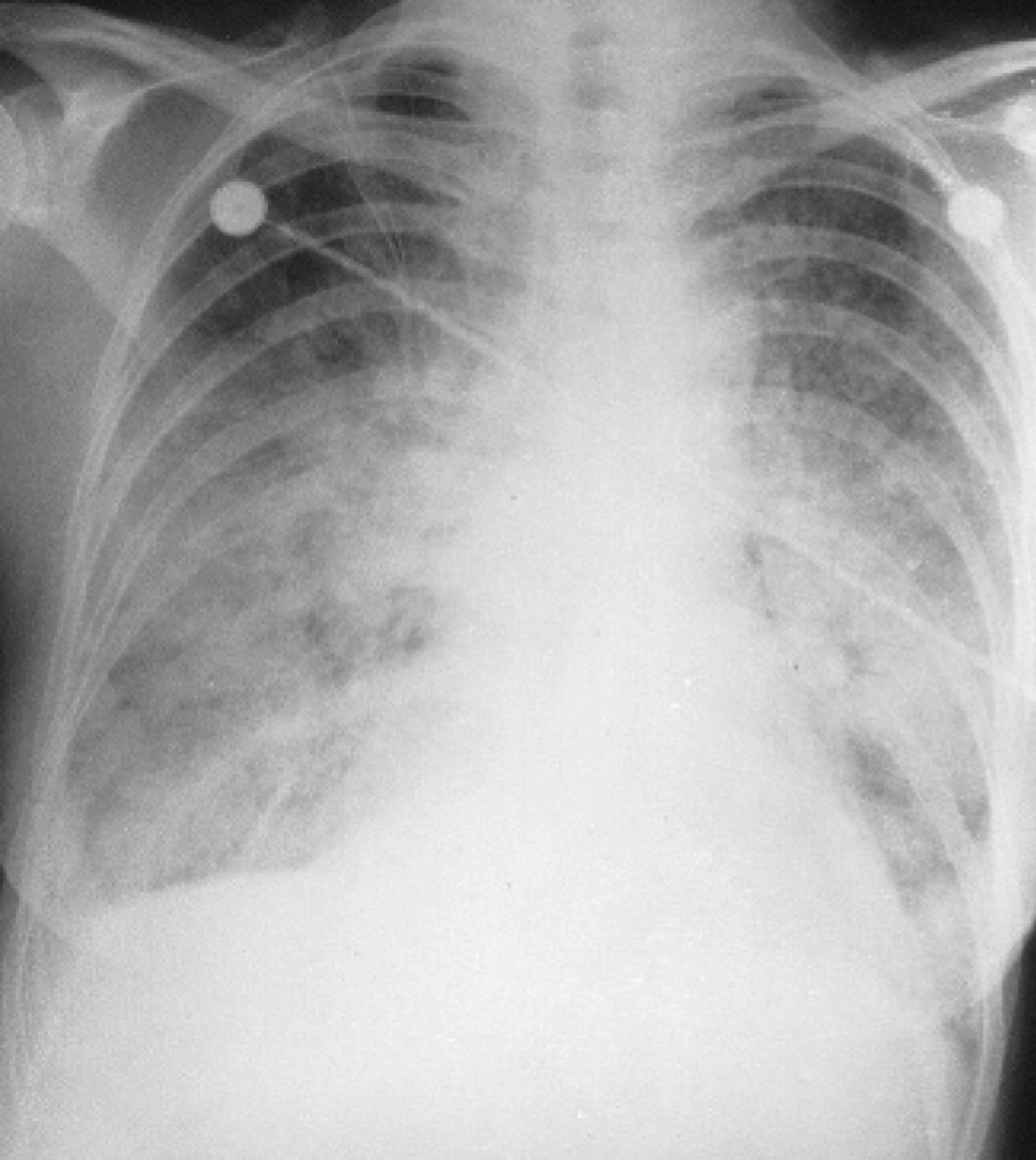
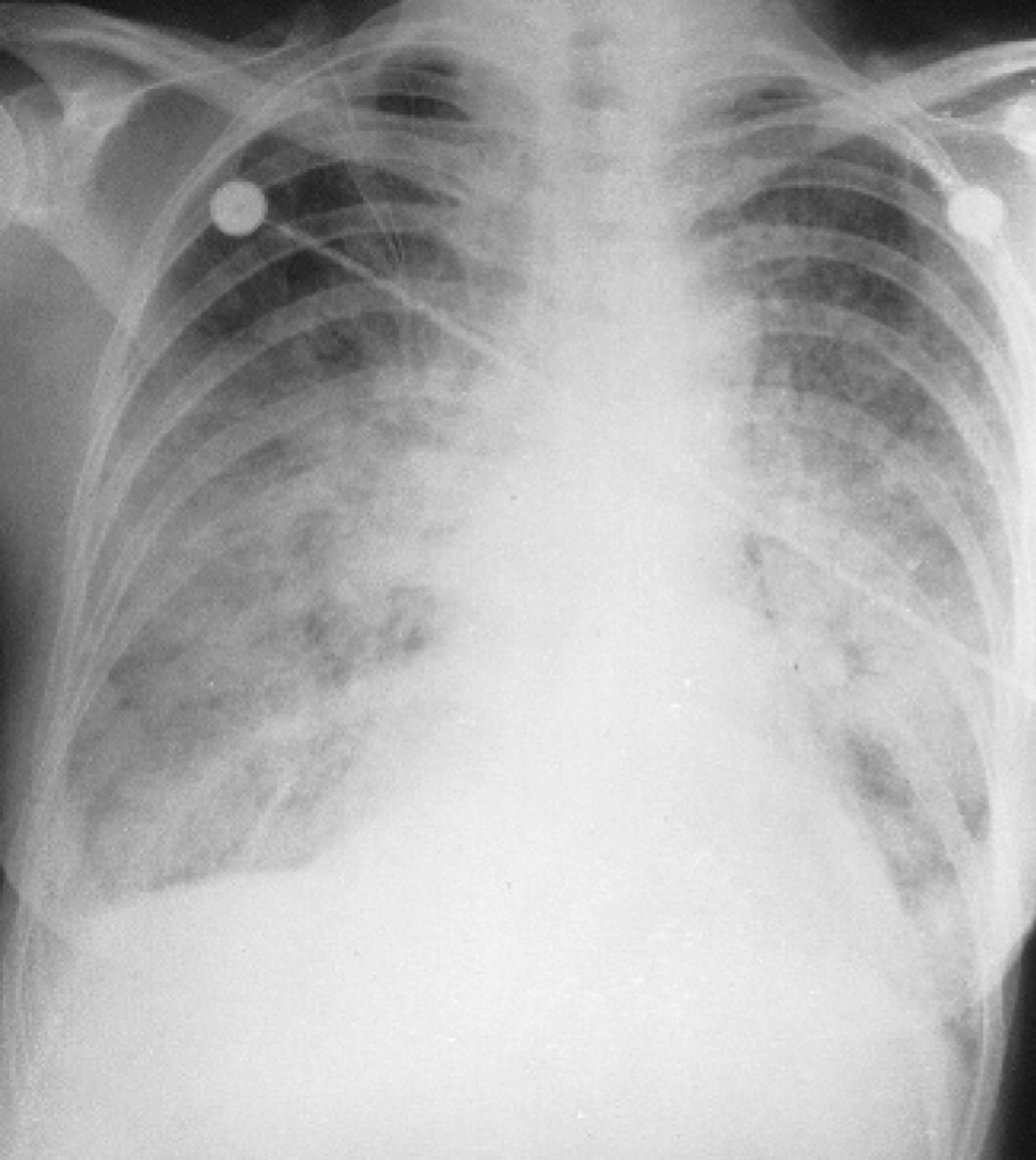
Acute pulmonary haemorrhage. Patient with vasculitis presenting with diffuse alveolar haemorrhage. Non-specific findings, with widespread interstitial opacification. The differential diagnosis includes infection, with opportunist infection possible. In such a case, early bronchoscopy with bronchoalveolar lavage should be considered once the patient's condition has been stabilised - however, transbronchial biopsy can be hazardous. A tissue diagnosis may have to wait open-lung biopsy.15 Reproduced with permission from Informa Healthcare.14
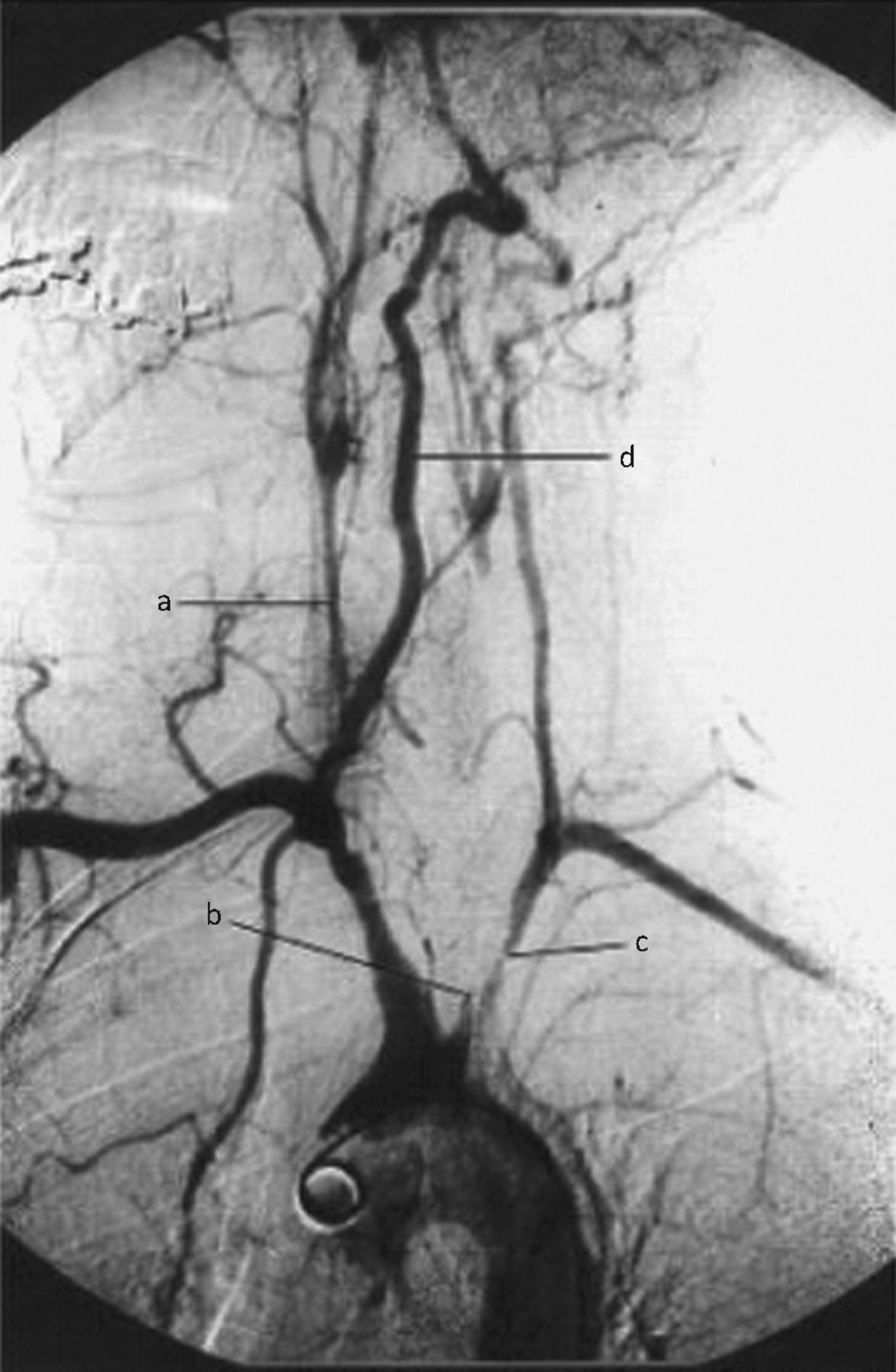
Magnetic resonance angiography (MRA) of a patient with Takayasu arteritis. Young female patient presenting with acute stroke, arterial bruits and discrepant upper limb blood pressures. Findings consistent with Takayasu arteritis: (a)= severely narrowed right common carotid artery; (b) = occlusion of left common carotid artery; (c)= proximal stenosis of left subclavian artery; (d) = right vertebral artery providing dominant cerebral supply. Reproduced with permission from BMJ Publishing Group Ltd.
Treatment
The principal questions in relation to treatment are:
Does the vasculitis require urgent treatment?
Is the vasculitis secondary to an underlying treatable cause?
Urgent treatment is required in the context of rapidly progressive glomerulonephritis in order to preserve the remaining renal function, and in temporal arteritis to reduce the risk of sudden blindness. Infection such as SBE or meningococcal infection requires urgent antibiotics in an attempt to minimise complications. If vasculitis is drug-induced, drug withdrawal may be sufficient. Immunosuppression may be required for patients with vital organ involvement, but the duration should be shorter than that in primary vasculitis. The prognosis is good provided that the offending drug is withdrawn in time.4
When faced with an acutely sick patient with signs consistent with either systemic infection or vasculitis, it can be difficult in the early stages to narrow the differential diagnosis. In some cases therefore, the combination of empirical immunosuppression and antibiotics can be justified pending further laboratory, imaging and histological information.
Recommended laboratory investigations for vasculitis.
Primary vasculitides
Treatment approaches for the primary vasculitides are considered in two phases: remission induction and subsequent maintenance.
A combination of steroid therapy with cyclophosphamide is often used for remission induction: intravenous (iv) methylprednisolone (500 mg-1 g daily for three doses) followed by oral prednisolone, or oral prednisolone (1 mg/ kg/day) with either oral cyclophosphamide (1.5–2 mg/kg/day) or pulsed iv cyclophosphamide (0.5–1 g/m2 body surface area/month for 3–6 months). Substitution of alternative steroid-sparing immunosuppression can then be considered to reduce the cumulative cyclophosphamide exposure.16–18
Plasma exchange, in combination with systemic immunosuppression can be considered in patients with vasculitis associated with pathogenic autoantibodies (eg WG19) and anti-GBM disease. It tends to be most effective when started as early as possible. Ideally, tests for such antibodies should therefore be available at all times, though this is rarely the case in practice.
The monoclonal antibody (MAb) rituximab, targeting the CD20 molecule on B cells, is being increasingly used in the context of vasculitis associated with pathogenic autoantibodies. It has been successfully applied to ANCA-associated vasculitis and cryoglobulinaemia.20,21 However, in view of the cost, MAbs are not currently first-line treatment.
High-dose iv Ig is the treatment of choice for Kawasaki disease, the greatest benefit being derived from early therapy. Low-dose aspirin is recommended for the thrombocythaemia. In large-vessel vasculitides, stenoses and/or aneurysm formation may require more aggressive intervention, either surgical or radiological, but the underlying disease process should be controlled in the first instance.
Other considerations for patients with primary vasculitides include mesna for those on iv cyclophosphamide, bone protection for those likely to be on long-term steroid, and infection prophylaxis while on immunosuppression.
Secondary vasculitides
Considerations for secondary vasculitides include:
drug withdrawal
appropriate treatment of infection (eg antibiotics in SBE and meningococcal disease, antiviral therapy for hepatitis C infection and HIV)
chemotherapy in the context of lymphoproliferative disease.
Acknowledgements
With thanks to Dr Bob Lock for the ANCA pictures, the Journal of Clinical Pathology and the Scandinavian Journal of Rheumatology for permission to use figures.
- © 2011 Royal College of Physicians
References








{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.