
Key points
Periodic fever syndromes (PFS) are very rare conditions resulting from innate immune dysregulation
Most PFS present in childhood with spontaneous febrile episodes which are associated with systemic and tissue inflammation
AA amyloidosis is the serious long-term complication found in a proportion of patients
Diagnosis can be challenging, and includes exclusion of other more common conditions such as occult infections, autoimmune diseases and malignancy
Routine genetic testing is available for the recognised PFS
Unexplained AA amyloidosis requires exclusion of PFS
Targeted anticytokine therapies are useful in management of PFS
Periodic fever syndromes are the founding members of a larger family of conditions known as autoinflammatory syndromes. Following the discovery of the genetic basis for TRAPS, the concept of autoinflammation was first proposed in 1999 to differentiate between the immunopathogenesis of PFS and that of classical autoimmune diseases.1 (See glossary on page 400 for explanation of abbreviations.)
The term autoinflammation has subsequently been expanded to encompass a number of immunologically mediated conditions that develop due to pathological inflammation mediated chiefly by the innate immune system. This is in contrast to autoimmune conditions where the disease process is principally due to aberrant adaptive immune responses and involvement of antigen-specific T and B cells. The appreciation that inflammation may be driven separately by either the innate or the adaptive immune system has led to some of the autoimmune diseases being reclassified as autoinflammatory conditions, whilst recognising that in a number of diseases with an intermediate phenotype there is a significant overlap.2 The discovery of the genetic causes of various PFS has transformed the approach to immunologically mediated inflammation and improved the understanding of those inflammatory diseases in which abnormalities of the innate immune system play a critical role. This review highlights the unique clinicopathological features of PFS in order to improve their recognition and diagnosis in everyday clinical practice.
Genetics and immunopathology of periodic fever syndromes
The phrase PFS usually refers to a group of hereditary periodic fevers which are inherited in either an autosomal recessive or dominant manner. They share some clinical features with other inherited autoinflammatory syndromes listed in Table 1. FMF is the most common and best characterised, and was the first PFS for which a genetic cause ? the presence of a mutation in the MEVF gene - was identified in 1997.3,4 The genetic bases of two further PFS were discovered in 1999:
TRAPS (previously known as Hibernian fever) was found to be due to a mutation affecting the TNFRSF1a gene1
HIDS is associated with a homozygous mutation in the MVK gene.5,6
Hereditary autoinflammatory syndromes: genetic and immunopathological features.
In 2001 a heterozygous mutation in the CISA1 gene was found to be responsible for FCAS and MWS.7,8 A year later a mutation in the same CISA1 gene was found also to cause CINCA/NOMID.9 Subsequently, the umbrella term CAPS was introduced to refer to these related clinical syndromes with a common genetic origin. In 2008 the first cases of periodic fever resembling FCAS, but associated with a mutation of NLRP12, were described.10
More recently, DIRA has been described. This condition is clinically distinct from CAPS, but with a similar immunopathological basis resulting from abnormalities in the IL-1 signalling pathway.11 DIRA is due to a homozygous mutation in the IL1RN gene and is classified as an autoinflammatory pyogenic disorder.
Common to all PFS is that genetic mutations in some way affect the processing of the important pro-inflammatory cytokines TNF and IL-1 which are produced principally by the innate immune system (Table 1). This is suggested not only by direct involvement of the mutated genes in IL-1 and TNF pathways but also by the beneficial effect of IL-1 and TNF blocking therapies in these conditions (Fig 1).12–14
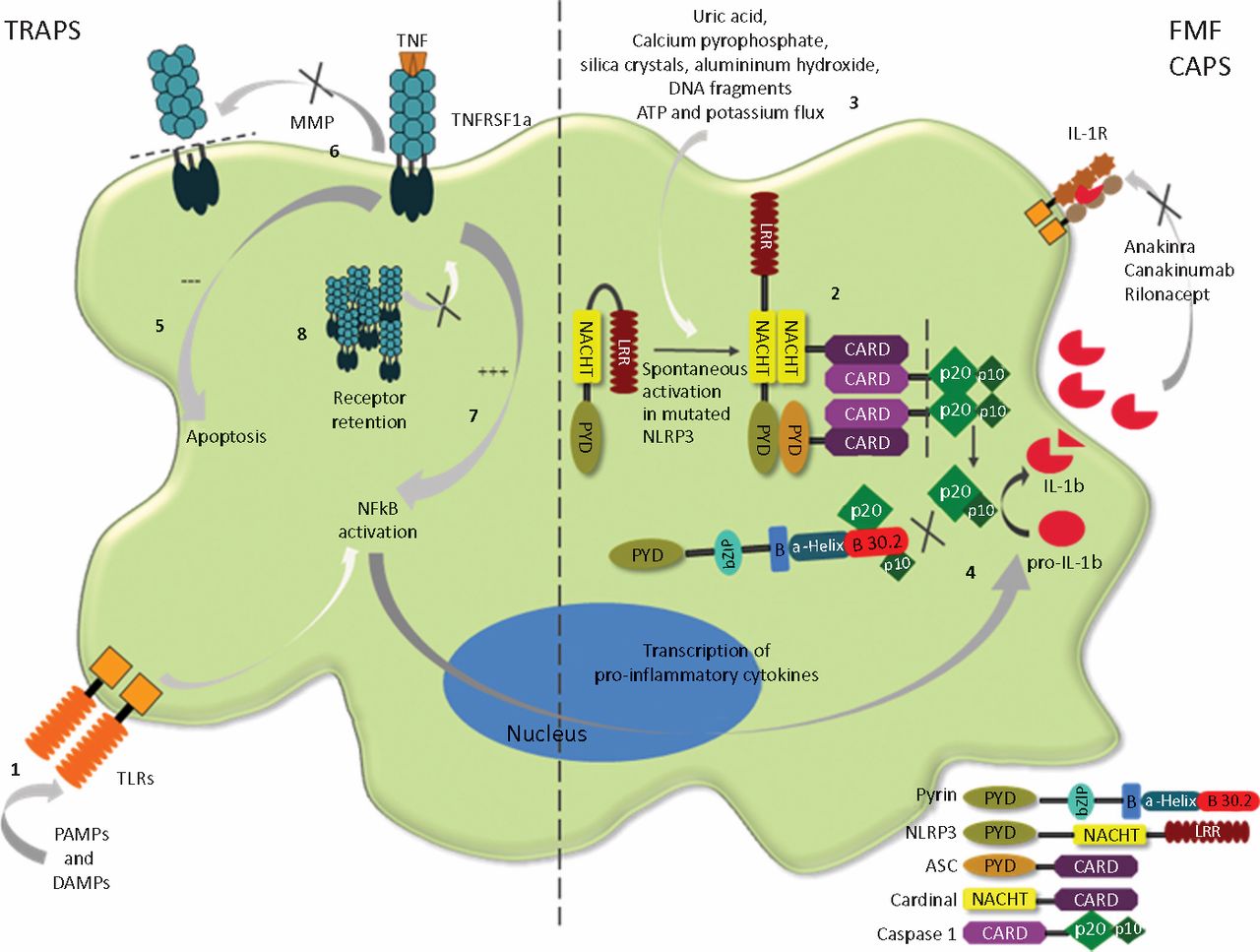
Immunology of CAPS and TRAPS. IL-1β secretion depends on two different pathways. First, signalling via TLRs (the structures of innate immunity which first recognise the presence of infection and tissue damage (PAMPs and DAMPs)), causes increased production of pro-IL-1β (1). This immature cytokine is then processed by a multimeric protein complex known as the NLRP3 inflammasome (2) which includes products of the MEFV gene (pyrin) and the CISA1 gene (cryopyrin). Activation of NLRP3 inflammasome normally depends on recognition of another set of ‘danger signals’ (3), which amongst others include DNA fragments, ATP and potassium flux, and which are indicative of tissue damage. Activated complex leads to autocatalytic activation of caspase −1, which then cleaves pro-IL-1β into the mature cytokine (4). Mutated cryopyrin appears to have a tendency towards self-activation leading to spontaneous production of mature IL-1β Pyrin, which normally limits caspase 1-mediated cleavage of pro-IL-1β by binding the catalytic subunits of caspase 1, p20 and p10, loses this ability in the mutated form. Excessive IL-1β effect is also seen in DIRA, which is due to the absence of natural IL-1β antagonists, IL-1ra (not shown). The effect of TNFRSF-1α gene mutation on TNFR1 signalling and the pathogenesis of TRAPS is less obvious. A number of different mechanisms have been suggested, including defective TNF-induced apoptosis (5), impaired receptor shedding leading to low levels of soluble TNFR1 (6), constitutive NF-κB activation (7),13 and impaired receptor trafficking (8).
Clinical features
Case history
PW is a 43-year-old Caucasian male who was well until his late teens. He then developed episodic attacks, characterised at first by abdominal and chest pain associated with fever and elevated inflammatory markers. There were no obvious triggers for these attacks, which would settle spontaneously after 7–10 days. Over the years he had an extensive number of investigations to exclude infective, malignant and autoimmune processes, some of them several times (eg abdominal and chest computed tomography scans). He also underwent an appendectomy and explorative laparoscopy, both of which were inconclusive.
In addition to his original problems, he developed a migratory erythematous rash associated with a painful underlying myalgia. Several empirical therapies were tried. The only effective treatment was systemic steroids such as prednisolone which shortened the duration of the attack when taken at the onset of typical symptoms. TRAPS remained undiagnosed for over 20 years. The only other family member found to be affected was his daughter, whose first recognised clinical presentation was at a similar age.
This case illustrates some of the clinical features classically associated with PFS as well as the diagnostic challenges such cases can present. Typically, spontaneous and episodic attacks of systemic inflammation occur, characterised by fever, serosal inflammation and varying degrees of muscular, articular, neurological and cutaneous involvement depending on the type of PFS (Table 2).15,16
Clinical features of periodic fever syndromes (PFS).
Most of these conditions will present in childhood but occasionally, as in this case, the first clear manifestations may be in early adulthood. They are classically episodic in nature, with periods of complete remission between attacks. However some, for example severe forms of CAPS, are associated with a more chronic course: in FMF and TRAPS there may be persistent subclinical inflammation17 between symptomatic episodes.
Aids to identification of different forms of periodic fever syndromes
A number of clinical characteristics can be helpful in identifying the different forms of PFS:
FMF tends to affect patients of Mediterranean ancestry and to present with an erythematous rash with the predilection for the lower limbs - unlike other PFS where the cutaneous involvement is more general.
HIDS almost always presents in childhood. Suggestive features include vomiting and diarrhoea associated with LAD and the triggering of attacks by immunisations.
Patients with TRAPS can present with characteristic periorbital oedema as well as myalgia, arthralgia, headaches and abdominal and chest pain.
FCAS, MWS and CINCA/NOMID share a number of features but retain certain distinctive clinical manifestations. Together, these define the disease continuum of CAPS, with FACS at the mild end, CINCA/NOMID at the severe end and MWS somewhere in the middle.
- Almost all CAPS patients will at some point have urticarial-like skin rash, with varying degrees of neurological muscoskeletal manifestations.
- In FCAS these symptoms are typically precipitated by exposure to cold and in addition to the skin rash, may also include conjunctivitis, fever and arthralgia.
- In MWS patients, these and a number of more severe manifestations such as arthritis and sensorineuronal deafness will usually occur spontaneously.
- CINCA/NOMID patients are the worst affected and tend to develop the characteristic complications outlined in Table 2.
AA amyloidosis (a form of amyloidosis associated with serum amyloid A protein) is a common serious complication affecting a proportion of PFS patients. Certain PFS are more frequently associated with amyloidosis than others (Table 2). Prevention of this complication necessitates early recognition and stringent control of systemic inflammation. Serum amyloid P scintigraphy can be used to detect amyloidosis early, whilst SAA levels can be used as a predictive marker for the development of tissue amyloidosis (levels ≤10 mg/l associated with significantly lower risk) as well as for monitoring treatment response. Where SAA amyloid measurement is not available, CRP can be used as a surrogate marker.
Investigations and differential diagnosis
The exact incidence of PFS in the UK is unknown but, with the exception of FMF which has a high prevalence in certain immigrant groups, these are very rare conditions. The diagnosis of PFS therefore is dependent on a high degree of clinical suspicion and, where possible, confirmation by genetic testing.
None of the routine investigations is particularly helpful in differentiating different PFS types. Elevated inflammatory markers (erythrocyte sedimentation rate, CRP and neutrophilia) are commonly found during the attacks but are non-specific markers. These may also be seen between episodes in otherwise asymptomatic patients, suggesting the persistence of subclinical inflammation. This is an important risk factor for developing systemic amyloidosis.
Serum IgD level measurement is of limited value in the diagnosis of HIDS because of the poor specificity and sensitivity of this test.18 Measurement of urinary mevalonic acid is potentially more useful as it tends to be elevated during an attack.
Other routine investigations should be directed towards excluding occult infections, malignancies and autoimmune diseases - far more common conditions typically considered in the differential diagnosis of PFS.
Important conditions which may mimic PFS, but which are not considered true PFS either because they lack a clear genetic basis or due to association with another pathology, are PFAPA and Schnitzler syndrome, respectively. PFAPA is usually a self-limiting paediatric illness, although adult cases have also been described recently.19 Schnitzler syndrome is a very rare disease found in adults, characterised by the presence of a monoclonal paraprotein (most commonly IgM isotype) but without features of lymphoproliferative disease. Patients tend to present with chronic urticaria, fevers, bone pain, LAD and hyperostosis.20
Various algorithms have been proposed to aid diagnosis of PFS. These can be helpful in alerting a physician to a possible diagnosis of PFS, but patients should be referred to a specialist with an interest in these conditions for further investigations and definitive diagnosis.
Treatment (Table 3)
Treatment options for periodic fever syndromes (PFS).
Most PFS respond to some extent to systemic steroids, but the response is frequently incomplete and a poor side effect profile precludes long-term use. Colchicine is uniquely and rapidly effective in FMF but has no place in the therapy of other PFS. This fact can help aid differential diagnosis.
Biologicals such as etanercept that block the function of TNF can be effective in the treatment of TRAPS. Note, however, that infliximab should be avoided as it can paradoxically cause a life-threatening flare of TRAPS.21 Anti-IL-1 blockade using any of a number of available biologics has not only transformed the treatment of CAPS but has also confirmed our understanding of the underlying immunopathogenesis. In addition to CAPS, selective IL-1 blockade is increasingly used in the therapy of HIDS and TRAPS.22
Acknowledgements
The authors would like to thank PW for giving consent for personal medical information used in this article.
Glossary
- AD
- autosomal dominant inheritance
- AR
- autosomal recessive inheritance
- ATP
- adenosine triphosphate
- CAPS
- cryopyrin-associated periodic fever syndrome
- CIAS
- cold-induced autoinflammatory syndrome
- CINCA
- chronic infantile neurologic, cutaneous and articular syndrome
- CRMO
- chronic recurrent multi focal osteomyelitis and congenital dyserythropoietic anaemia
- CRP
- C-reactive protein
- DAMP
- damage-associated molecular pattern
- DIRA
- deficiency of interleukin-1 receptor antagonists
- FCAS
- familial cold autoinflammatory syndrome
- FMF
- familial Mediterranean fever
- HIDS
- hyperimmunoglobulinaemia D with period fever syndrome
- Ig
- immunoglobulin
- IL
- interleukin
- LAD
- lymphadenopathy
- MVK
- mevalonate kinase
- MWS
- Muckle-Wells syndrome
- NAPS12
- NLRP12-associated periodic syndrome
- NF
- nuclear factor
- NLRP3
- nucleotide-binding domain, leucine-rich repeat containing protein (NLR) family, pyrin domain containing 3
- NOD
- nucleotide-binding oligomerisation
- NOMID
- neonatal onset multisystem inflammatory disease
- PAMP
- pathogen-associated molecular pattern
- PAPA
- pyogenic sterile arthritis, pyoderma gangrenosum, acne
- PFAPA
- periodic fever, aphthous stomatitis, pharyngitis, adenopathy
- PFS
- periodic fever syndrome
- SAA
- serum amyloid
- TLR
- Toll-like receptor
- TNF
- tumour necrosis factor
- TNFRSF1A
- tumour necrosis factor receptor superfamily member 1A
- TRAPS
- tumour necrosis factor-associated periodic fever syndrome
- © 2011 Royal College of Physicians
References








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.