
Abstract
Autoimmune limbic encephalitis is an increasingly recognised cause of cognitive decline and confusion. The typical presentation is with subacute cognitive decline, behavioural disturbance and seizures. Magnetic resonance imaging may show characteristic changes in the medial temporal regions. The diagnosis is confirmed by identification of elevated voltage-gated potassium channel antibody (VGKC-Ab) titres. It is a highly treatable condition, often responding well to intravenous immunoglobulin or steroids. Recognition of autoimmune limbic encephalitis is sometimes delayed–usually because the diagnosis has not been considered–which can result in long-term neurological consequences.
Key learning points
Autoimmune limbic encephalitis is an increasingly recognised cause of subacute cognitive decline, often associated with seizures and hyponatraemia.
Characteristic changes are often seen on magnetic resonance imaging, and titres of voltage-gated potassium channel antibodies are typically elevated.
Early consideration of the diagnosis is vital, as delayed treatment may be associated with worse neurological outcome.
Treatment is with intravenous immunoglobulin and cortiosteroids, and is often highly effective.
Case history
A 58-year-old man presented to the emergency department with four generalised tonic-clonic seizures. His family reported that he had become increasingly withdrawn over the preceding year with intermittent behavioural change and confusion, and occasional visual and auditory hallucinations. He had reported seeing his mother (who had died 11 years previously), and hearing voices coming from a plug socket. Between episodes he had been lucid and orientated. He had been admitted with idiopathic pancreatitis in the previous month; he also had a history of myocardial infarction four years previously, and had fractured his skull in a fall many years ago. He had no known history of psychiatric illness or epilepsy. On examination, he was confused and drowsy. He was afebrile with no meningism. General and neurological examination was otherwise normal.
What is the differential diagnosis and the most likely diagnosis?
It is important to distinguish between symptomatic seizures, with an identifiable cause, and unprovoked seizures (ie epilepsy). Possible causes of symptomatic seizures here include: metabolic disturbance; brain pathology, such as tumour or stroke; infection, such as herpes simplex encephalitis; or a toxic cause, such as alcohol withdrawal.
The longer history of fluctuating behavioural change must also be considered. This could be due to a dementia, particularly dementia with Lewy bodies or Alzheimer's disease. An underlying psychiatric condition, such as schizophrenia, should be considered.
With seizures and behavioural change, a frontal tumour (either primary or secondary) is plausible; the relatively long period of cognitive change might suggest a slower growing tumour, such as a meningioma. Alternatively, more than one condition may be present; for example, he might have an underlying dementia, with seizures and delirium precipitated by an acute event. A recurrence of pancreatitis might be such a precipitant here (particularly if associated with electrolyte imbalance); this might also suggest unrecognised heavy alcohol consumption.
Less common neurological conditions should also be considered; a history of behavioural and cognitive change associated with seizures raises the possibility of limbic encephalitis or Hashimoto's encephalopathy.
A full differential diagnosis is given in Table 1.
The differential diagnosis of limbic encephalitis.
What is the initial management?
Initial management is aimed at terminating seizures, and identifying and treating any underlying cause. Four seizures (without full recovery in between), occurring over a period of more than 30 minutes, meet the criteria for status epilepticus and should be treated accordingly. Blood should be sent urgently for blood glucose levels, biochemistry (including urea and electrolytes, calcium, magnesium), and haematology. Alcohol levels and toxicology screen may be indicated. Neuroimaging with computed tomography (CT) is required initially, and if central nervous system infection is suspected a lumbar puncture should subsequently be performed.
Immediate treatment should be with intravenous (iv) lorazepam, followed by iv phenytoin loading if seizures do not settle; transfer to intensive care for anaesthesia, intubation and ventilation may be required. Further specific treatments depend upon investigation findings and clinical progression.
Case progression
Initial investigations revealed hyponatraemia (120 mmol/l), serum amylase 400 U/l (normal range <12 U/l). CT abdomen showed a pancreatic pseudocyst and CT head showed an old left parietal skull fracture. Brain magnetic resonance imaging (MRI) showed increased signal in the right medial temporal gyrus on T2-weighted sequences, initially attributed to previous trauma or ischaemia. Cerebrospinal fluid (CSF) analysis revealed mildly elevated protein (0.52 g/l, reference range 0.15–0.45 g/l), but was otherwise normal.
His presentation was attributed to metabolic disturbance from recurrent pancreatitis on the background of a previous head injury. His seizures settled on a combination of phenytoin and sodium valproate, and serum sodium normalised. However, his confusion and hallucinations progressively worsened. Seven weeks after admission, he had an upper gastrointestinal haemorrhage, and was transferred to the regional gastroenterology centre. Transfer was complicated by status epilepticus necessitating intubation and admission to the intensive care unit. His conscious level did not improve on reduction of sedation, and an electroencephalogram (EEG) showed focal nonconvulsive status epilepticus. Repeat MRI showed worsening of the previously seen temporal lobe abnormalities, with high signal in both hippocampi (Fig 1) and involvement of the right temporal cortex on T2-weighted sequences. Limbic encephalitis was suspected, and subsequently confirmed with VGKC-Ab markedly elevated at 958 pM (reference range <100 pm).
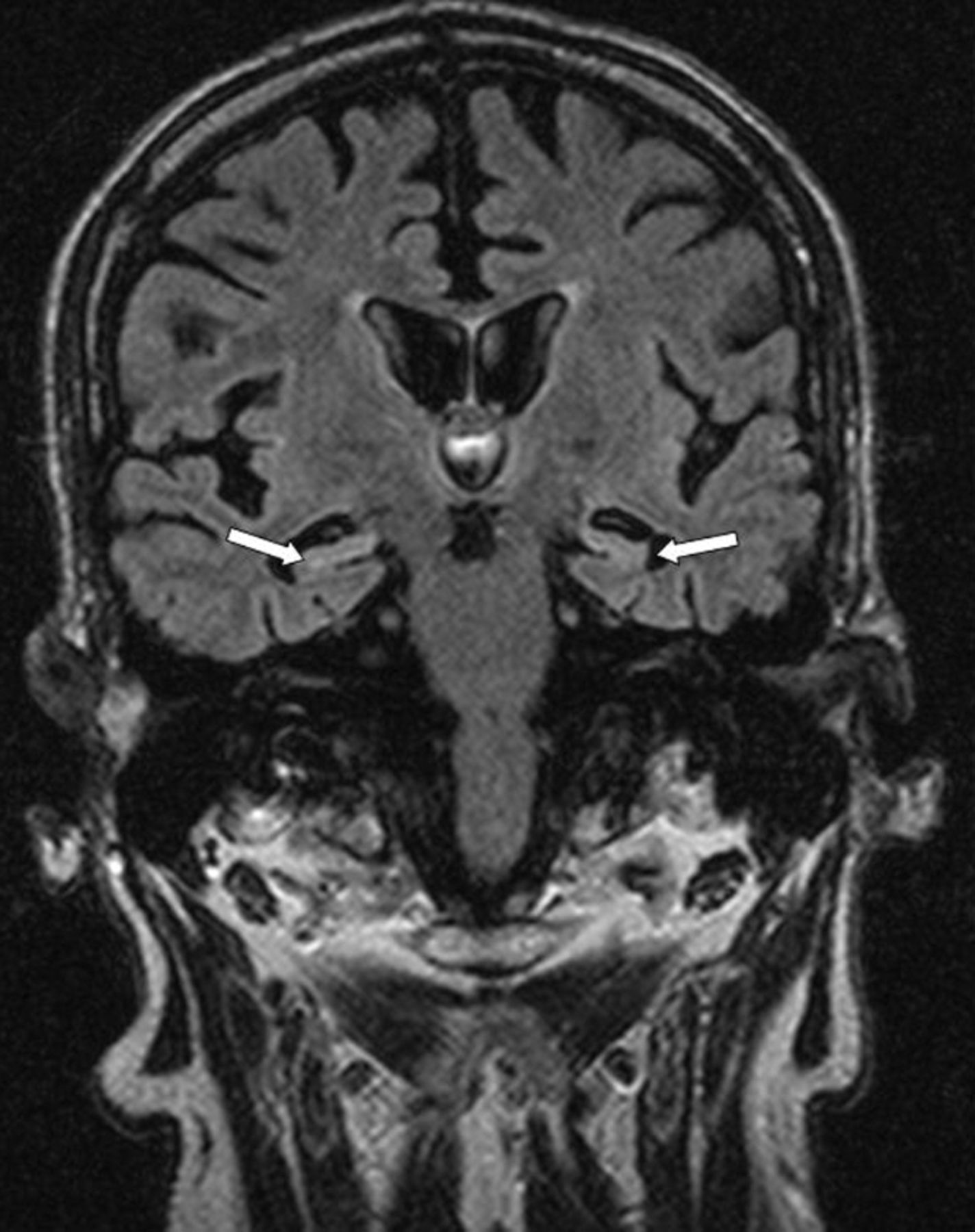
Magnetic resonance image (FLAIR sequence), showing generalised cerebral atrophy and relative hyperintensity in medial temporal structures (arrowed).
At this point he was treated with iv immunoglobulin (IVIG) but remained comatose off all sedation. A course of high dose iv methylprednisolone, followed by a five-day course of plasma exchange, resulted in no improvement. He developed bronchopneumonia and died three months after admission. Post mortem examination of the brain showed striking neuronal loss and gliosis in the temporal and frontal lobes, with minor lymphocytic infiltrate. The pathological findings confirmed relatively inactive limbic encephalitis, with no evidence of underlying malignancy anywhere in the body.
Discussion
Limbic encephalitis (LE) is an increasingly recognised, potentially treatable condition which is often misdiagnosed as dementia or delirium; diagnostic delay is common and may lead to worse outcomes.
Clinical features
LE typically presents as a subacute amnestic syndrome, usually evolving over weeks to months. Memory problems, with periods of confusion and disorientation, are common; intellectual decline and psychiatric features (including hallucinations and personality change) may occur.1,2 Focal and secondarily generalised seizures are common. Neurological examination is usually unremarkable, but global impairments of verbal and visual memory are typically present on neuropsychological assessment.2
Initial investigations, including CT brain imaging, may be normal, although hyponatraemia is common.2 MRI may show hyperintense temporal lobe signal changes on T2 and FLAIR sequences, sometimes progressing to hippocampal and temporal lobe atrophy.1–4 The MR findings are important, and were an important clue in this case, but are not always present. Analysis of the cerebrospinal fluid may show a lymphocytic pleocytosis, mildly elevated protein, and oligoclonal bands, but is often normal.1–3 EEG typically shows either focal or generalised slow wave abnormalities, sometimes with epileptiform features in the temporal regions.1,2 Histopathology is rarely available in life, but characteristically shows inflammatory changes in limbic structures, particularly the hippocampus and amygdala.1
LE occurs in two forms, which are difficult to distinguish clinically; a paraneoplastic form, most commonly occurring in association with small-cell lung cancer, and often associated with antineuronal antibodies (particularly anti-Hu),1,5 and an autoimmune form, associated with VGKC-Ab.2,3 While paraneoplastic LE generally has a poor prognosis, autoimmune LE may respond dramatically to immunomodulatory therapy.2,3 It is likely that in autoimmune LE, the VGKC abs are pathogenic although recent work has indicated that the antigenic target is not in fact the potassium channel.6
As paraneoplastic LE may antedate the diagnosis of cancer in about 60–80% of cases,1,5 screening for malignancy is indicated. VGKC-Ab are rarely present in paraneoplastic limbic encephalitis, although low titres (< 400 pM) may occur.7 VGKC-Ab negative autoimmune limbic encephalitis may occur, and other antibodies are increasingly recognised in association with these cases.8,9
Treatment and prognosis
Treatment of paraneoplastic LE is focused on the underlying malignancy; immunosuppression is largely ineffective and the prognosis is poor, although there are occasional good outcomes.1
In contrast, autoimmune LE often responds well to immunotherapy.2,3,9 Although there are few data on which to base management, most authorities recommend a combination of IVIG or plasma exchange with high dose steroids10; substantial clinical improvement is often seen following treatment.2,3 Immunotherapy is usually continued for a period of months and gradually tapered, with doses titrated against clinical state and VGKC-Ab titre which tends to fall in parallel with clinical improvement.2,10 Improvement may continue for months after treatment begins. However, permanent deficits remain in a proportion of patients,2,3 probably reflecting permanent cerebral damage, ongoing inflammation resistant to treatment, or a combination of both. Although robust evidence is lacking, earlier treatment appears to result in better outcome2,10; untreated, cerebral inflammation, frequent seizures and sometimes status epilepticus associated with LE result in irreversible neuronal damage and occasionally death.
Funding
RASS is funded by a Medical Research Council senior clinical fellowship.
- © 2011 Royal College of Physicians








{kind=link}