
Key Points
‘Red flag’ features warrant further investigation
A high fluid intake and vegetarian diet are important preventative measures for the majority of recurrent stone–formers
Further treatment is guided by urine chemistry
The presence of an underlying condition should be considered in patients with pure calcium phosphate or uric acid stones
Enteric hyperoxaluria, primary hyperoxaluria, and cystinuria are ideally managed in renal or metabolic stone clinics
Renal colic accounts for about 1% of hospital admissions worldwide and is the reason for 80,000 emergency department visits per year in the UK. The initial episode is normally dealt with by urologists, but physicians are increasingly encountering patients with nephrolithiasis because of its association with hypertension, obesity, diabetes and osteoporosis.
Epidemiology
Kidney stone disease typically presents between the ages of 20 and 60 and is more prevalent in hot climates.1 It affects about 10% of people over their lifetime, incidence increasing with age; 50% will have a recurrence within 5–10 years and 75% within 20 years.2 Developed countries have seen rapid increases over the last 30 years, especially in women in whom incidence is now almost equal to that of men.3
This article focuses on the pathophysiology, investigation and management of recurrent stone disease.
Pathophysiology
Stone growth starts with the formation of crystals in supersaturated urine which then adhere to the urothelium, thus creating the nidus for subsequent stone growth. The biological processes that anchor crystals to the urothelium are incompletely understood. Many, but not all, calcium oxalate stones develop on Randall's plaques which are composed of calcium phosphate (= hydroxyapatite) crystals. These grow to erode the urothelium, forming a nucleus for calcium oxalate deposition.
More recent theories focus on the role of cell surface molecules which favour or inhibit crystal adhesion.4,5 Urothelial injury and repair after a stone episode may increase surface expression of these molecules to favour further crystal adhesion.6 Thus ‘stones beget stones’7 because there may be a residual nucleus on which further stones may form and/or upregulation of molecules favouring crystal adhesion. Stone prevention focuses on identifying and ameliorating the risk factors for crystal formation.
Risk factors
Low fluid intake
The single most important determinant of stone formation is low fluid intake. A low fluid intake results in the production of concentrated urine, causing supersaturation and crystallisation of stone–forming compounds. In addition, low urine flow rates favour crystal deposition on the urothelium.
Hypercalciuria
About 80% of stones are calcium based, predominantly either calcium oxalate (70%) or calcium phosphate (10%). High urine calcium is the single most common abnormality of urine chemistry in recurrent stone formers, but until recently the relative contributions of altered gut absorption, bone turnover, and renal handling were poorly understood. US texts promote the concept that hypercalciuria can be divided into ‘absorptive’ and ‘renal’ phenotypes, but there is scant evidence that these phenotypes are reproducible or that different therapeutic approaches are justified in patients with different phenotypes.8 An appreciation of the mechanisms of calcium homeostasis helps to understand how hypercalciuria may develop (Fig 1).
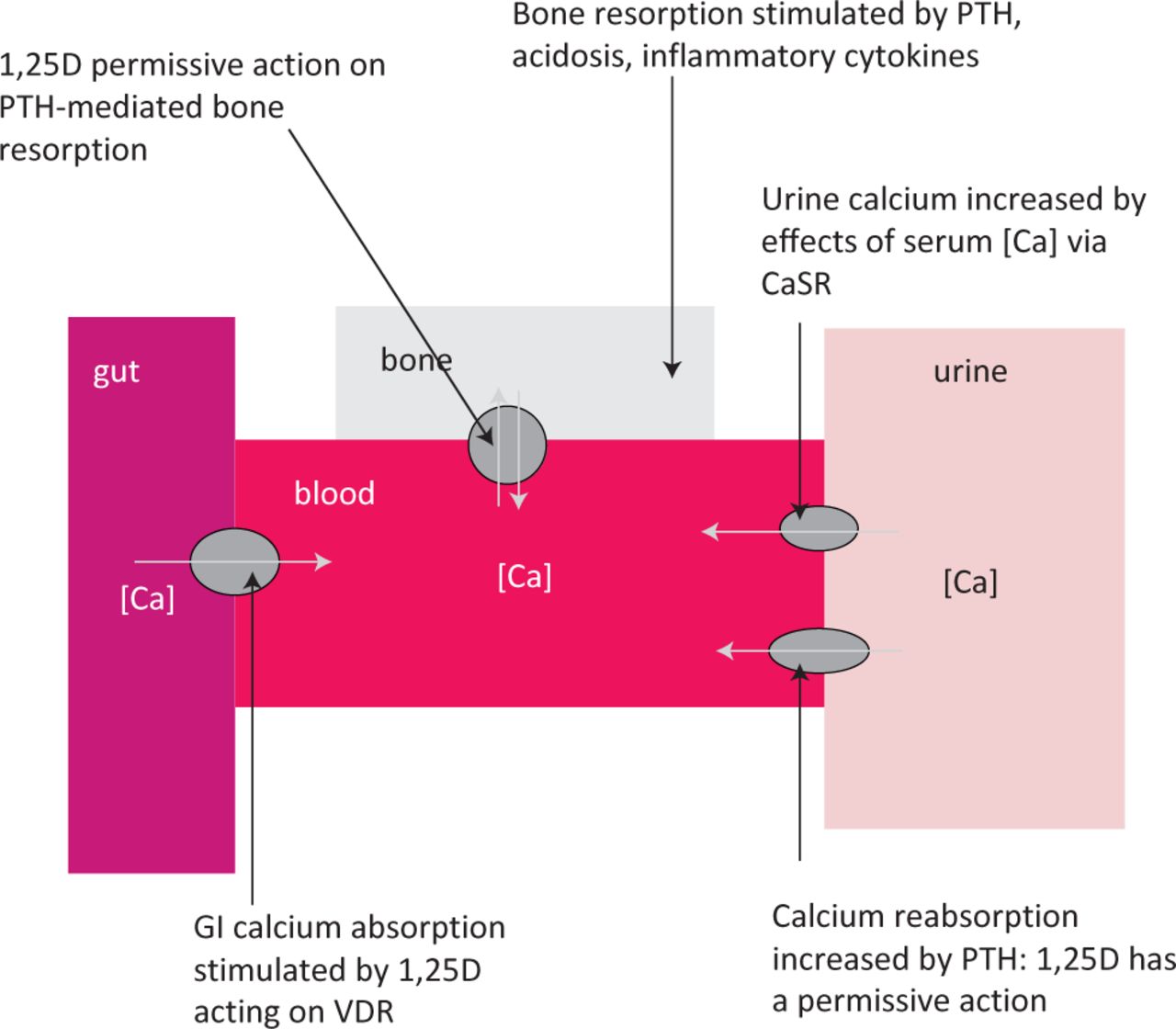
Determinants of urine calcium excretion. Increased urinary calcium excretion can be caused by increased gastrointestional absorption, increased bone resorption or increased tubular reabsorption. All three processes are influenced by a complex series of feedback loops with PTH and 1,25D being the major hormones involved. 1,25D acts via the vitamin D receptor. 97% of filtered calcium is normally reabsorbed, so urine calcium excretion is controlled by changes in reabsorption. A rise in serum calcium increases calcium excretion by acting on the CaSR. Calcium reabsorption is stimulated by PTH, with 1,25D playing a permissive role. High sodium chloride intake also reduces renal tubular calcium reabsorption (not shown). Decreased renal tubular phosphate reabsorption may also indirectly affect calcium excretion by stimulating increased 1,25D. PTH is increased by low serum [Ca], low serum [1,25D], high serum [PO4] and low serum [FGF23]. 1,25D is increased by PTH and low serum [PO4]. 1,25D = 1,25 dihydroxyvitamin D; CaSR = extracellular calcium receptor; [Ca] = calcium; FGF23 = fibroblast growth factor; PO4 = phosphate; PTH = parathyroid hormone.
An underlying genetic basis for ‘idiopathic’ hypercalciuria is evident from the observation that high urine calcium is the most consistent difference between stone–formers with and without a family history of stones.9–11
Hypercalciuria and an excess risk of stone formation is seen in patients with primary hyperparathyroidism, deactivating vitamin D receptor (VDR) polymorphisms and activating fibroblast growth factor (FGF) 23 polymorphism.12,13
Deactivating VDR variants
The development of stone disease is likely to occur only in the presence of additional risk factors. For example, patients with deactivating VDR variants form stones if there is associated hypocitraturia. VDR activity promotes citrate excretion14 which increases the solubility of calcium salts. A diet low in fruit and vegetables (which also boosts citrate excretion) may predispose to stone formation in these patients. Citrate supplementation may be a particularly effective therapeutic intervention.
Primary hyperparathyroidism
In primary hyperparathyroidism, activated extracellular calcium–sensing receptor (CaSR) causes urinary dilution and acidification, protecting against stone formation by preventing supersaturation of urine with insoluble calcium salts. Stones develop in hyperparathyroid patients with allelic variants that cause reduced expression of the CaSR, with associated loss of its urinary dilution and acidification effect.15
High salt diet
A high salt diet increases urinary calcium output.16 The sodium/chloride co–transporter at the loop of Henle drives the electric gradient required for paracellular calcium uptake. Sodium bicarbonate does not cause hypercalciuria, provided that dietary chloride is kept low,17 suggesting that it is chloride rather than sodium that determines calcium uptake. In one trial a reduced salt intake was one component of a successful intervention to reduce nephrolithiasis rates.18 However, a high salt intake may also help to increase spontaneous water intake, offsetting the effect on calcium excretion.19
Factors contributing to high urine oxalate
Most stones are calcium oxalate. There are very high urine oxalate levels in patients with both primary and enteric oxaluria (see below). No underlying cause is identified in most calcium oxalate stone–formers and urine oxalate is often within the reference range. Intestinal oxalate absorption is higher on average in stone-formers than in non–stone– formers: it is possible that intestinal oxalate transporter polymorphisms contribute to the risk of calcium oxalate stone formation.20 Dietary modification leading to small reductions in urinary oxalate, even within the reference range, can significantly reduce the risk of calcium oxalate stones.21
Low calcium intake
The inverse association between dietary calcium intake and stone formation seems counterintuitive. In fact, the association is proven,22,23 reflecting the ability of dietary calcium to limit intestinal oxalate absorption.18
Hypocitraturia
Citrate reduces calcium activity in the urine by forming soluble complexes with calcium and is an important inhibitor of crystallisation. Its excretion is partly determined by filtered load of citrate and partly by systemic acid–base balance. Hypocitraturia is found in hypokalaemia, chronic acidosis (including that caused by ileostomy diarrhoea) and in distal renal tubular acidosis.24
High animal protein intake
Animal protein (meat, poultry, fish) is metabolised to oxalate and uric acid. Uric acid causes nucleation of calcium oxalate crystals. Metabolism of animal protein generates fixed acid, reducing intake of animal protein lowers urine oxalate and raises urine pH, reducing the risk of both oxalate and uric acid stones.25,26
Enteric hyperoxaluria
Patients with short bowel syndrome and a functioning colon develop hyperoxaluria (‘enteric hyperoxaluria’) and kidney stones. In this condition, increased colonic absorption of oxalate occurs by a combination of decreased luminal calcium activity (as the result of calcium binding to unabsorbed fatty acids) and increased colonic permeability to oxalate (caused by unabsorbed bile salts).
Primary hyperoxaluria
Endogenous oxalate metabolism is a key determinant of urinary oxalate. The primary hyperoxalurias (PH) are a paradigm for the consequences of disturbed oxalate metabolism. They are autosomal recessive conditions characterised by urine oxalate output typically exceeding 800 μmol/24 hours, subtyped according to the affected enzyme: PH1 is the most severe and PH3 the least. Genetic analysis distinguishes subtypes.
In a milder variant of PH1, there is mis–targeting rather than complete absence of the enzyme. Patients are responsive to high–dose pyridoxine, a co–factor for the enzyme. It is likely that other genetic polymorphisms affecting oxalate metabolism will be identified.
Investigations
Not all patients with kidney stones should be investigated with a view to defining the underlying abnormality. After a single stone episode, many patients would not be convinced of the need to modify their lifestyle or take regular drug treatment to avoid the risk of recurrence. The presence of a number of ‘red flag’ features warrants further investigations being offered (Box 1).
‘Red flag’ features warranting further investigation.

A detailed investigation algorithm is available in a recent review.27 The minimum initial investigations we recommend are outlined in Table 1. Diurnal calcium excretion is highly variable. Ideally, therefore, patients should provide two 24–hour urine collections. If urine oxalate output exceeds 800 μmol/24 hours, genetic analysis for PH is advised. If cystinuria is suspected and stone analysis not available, a spot urine sample for urine amino acids (cystine concentration) should be collected.
Initial investigations in patients with ‘red flag’ features.
Analysis of the biochemical composition of kidney stones, either passed spontaneously or retrieved during percutaneous or endoscopic surgery, is extremely valuable in directing further investigation and treatment. Where this is not available, radiology may provide some clues – uric acid and cystine stones are characteristically radiolucent on plain radiology. Stone density (Hounsfield units) on unenhanced computed tomography may be helpful for larger stones. Additional investigations may be indicated in non–calcium oxalate stone formers (see below).
Calcium phosphate stones
Targeted additional investigations for calcium phosphate stone–formers are summarised in Table 2. If distal renal tubular acidosis (dRTA) is identified,28,29 an underlying cause should be sought. Calcium phosphate stones are a feature of many rare monogenic diseases30 including inherited forms of dRTA and the X–linked condition, Dent's disease.
Targeted investigations for calcium phosphate stone–formers (CT 5 computed tomography).
Urine phosphate
Measurement of urine phosphate is of no proven value in the diagnosis or management of stone formers. In hyperphosphaturic patients, the risk of stone formation is determined by urine pH. Relatively soluble monocalcium phosphate (Ca(H2PO4)2) salts form at pH 6, whilst at pH 8 insoluble dicalcium phosphate (CaHPO4) salts form. Hence, alkaline urine favours formation of calcium phosphate stones.
High urinary phosphate excretion is normally associated with low urinary pH because most dietary phosphate is acidic. The presence of other cations such as sodium and potassium, which form soluble phosphate salts in alkaline urine, may also determine the extent to which insoluble calcium phosphate salts form at high urine pH.
Uric acid stones
Uric acid stones, or mixed uric acid and calcium oxalate stones, are most commonly seen in patients with concentrated acidic urine and features of metabolic syndrome. Measurement of serum urate, glycated haemoglobin (HbA1c) and blood pressure should form part of the investigation. Patients with an ileostomy are at risk of uric acid stones because of high bicarbonate and fluid losses. Increased cell turnover, occurring for example in myeloproliferative disorder and inflammatory bowel disease, is also associated with uric acid stones. Screening for myeloproliferative disorder is with a full blood count. Measurement of urinary uric acid output is unhelpful because the solubility of uric acid in urine is highly pH–dependent: at low pH, even small amounts will precipitate, whereas the reverse is true at high pH.31
Cystine stones
The only situation in which cystine stones occur is cystinuria, an autosomal recessive condition caused by abnormal transport of dibasic amino acids, including cystine. Affected individuals experience recurrent stone formation from a young age and may ultimately develop kidney failure.
Management
All patients in whom further management is appropriate should receive dietary and lifestyle advice. In temperate climates, a fluid intake of at least two litres a day halves recurrence rates.32 A diet high in fruit and vegetables is recommended because the high potassium content promotes urinary citrate excretion. These foods are also a source of phytates which, like citrate, increase calcium salt solubility. An adequate calcium intake, with restricted animal protein, reduces urine oxalate. A limited salt and sugar intake is also advised. Where possible, an underlying disorder predisposing to stone formation should be identified and treated.
Further treatment
Additional treatment should be guided by urine chemistry (Table 3):
Thiazide diuretics reduce urinary calcium and halve stone risk in hypercalciuric patients.33
The addition of amiloride may boost citrate excretion by its potassium–sparing effect.
Potassium citrate is indicated in hypocitraturia and is also used as a urine alkalinising agent.
Urine alkalinisation with citrate or bicarbonate increases the solubility of uric acid, cystine and calcium oxalate stones. Doses are titrated to achieve an ideal urine pH 7. A higher pH exposes patients to the risk of calcium phosphate stones, particularly in the presence of hypercalciuria.
In enteric hyperoxaluria the mainstay of treatment is rigorous dietary restriction of oxalate combined with an adequate calcium intake.
As in all types of kidney stone disease, a high volume dilute urine is desirable in enteric hyperoxaluria. However, in patients with short bowel, a high water intake can exacerbate diarrhoea without improving urine dilution. A solution containing electrolytes and glucose (eg St Mark's solution) may be preferable to plain water.
The administration of ‘oxalate binders’, analogous to the use of phosphate binders in chronic kidney disease, is logical but clinical studies proving benefit in enteric hyperoxaluria have not been reported.
Struvite (triple phosphate) stones are the staghorn calculi associated with urinary tract infection and precipitate in alkaline urine. Treatment is with antibiotics and stone clearance.
Abnormalities of urine chemistry that increase risk of kidney stones and warrant further investigation (PH = primary hyperoxaluria).
Management in nephrology or metabolic stone clinics
Both enteric hyperoxaluria and PH are ideally managed in nephrology or metabolic stone clinics because they can lead to progressive renal failure as a consequence of kidney stones, their treatment and oxalate deposition within the kidney. End–stage renal failure can be complicated by systemic oxalosis and multi–organ failure.
In cystinuria, cystine–binding drugs may be indicated if stones recur despite conservative measures and urine alkalinisation. This should ideally be monitored in a nephrology or metabolic stone clinic.
Conclusions and future work
The great majority of recurrent stone formers do not have a recognised underlying cause for their condition. A careful history may reveal a number of dietary and lifestyle risk factors contributing to an individual's risk of stone disease. A more complete understanding of the influence of dietary and lifestyle factors on the phenotypic expression of genetic polymorphisms which predispose to stone formation may facilitate a more tailored approach to stone prevention in the future.
- © 2012 Royal College of Physicians
References








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.