
- renal tubule
- proximal tubule loop of Henle
- collecting duct
- renal Fanconi syndrome
- renal tubular acidosis
Key points
Disorders of the proximal tubule can cause renal Fanconi syndrome with glycosuria, amino aciduria, bicarbonaturia, phosphaturia (often, though not always, with hypercalciuria), uricosuria, and low molecular weight tubular proteinuria (not usually detectable by routine urine dipstick)
Bartter syndrome is caused by mutations that inactivate the loop diuretic-sensitive NKCC2 cotransporter in the thick ascending limb of Henle's loop. The electrolyte abnormalities found in Bartter syndrome are similar to those occurring on loop diuretics
Gitelman syndrome is caused by mutations that inactivate the thiazide diuretic-sensitive NCC cotransporter. The electrolyte abnormalities of Gitelman syndrome are similar to those occurring on thiazide diuretics
Genetic causes of hypertension can result from activating mutations of NCC (Gordon syndrome) or of ENaC (Liddle syndrome). They are a ‘mirror image’ of Gitelman syndrome and pseudohypoaldosteronism type 1a, respectively
Proximal (tubular) RTA is caused by failure to reabsorb bicarbonate and is usually part of the renal Fanconi syndrome. Whereas distal (tubular) RTA is caused by failure to secrete H+, proximal (tubular) RTA is often associated with autoimmune disease in adults and causes a more severe form of acidosis with hypokalaemia, complicated by stones and nephrocalcinosis
The physiology of the renal tubule and the diseases that can affect its function are often thought of as complicated and confusing. This article will attempt to make tubular disorders slightly easier to understand by linking the physiology of the four main nephron segments with the clinical features of the more commonly encountered renal tubular disorders (Fig 1).
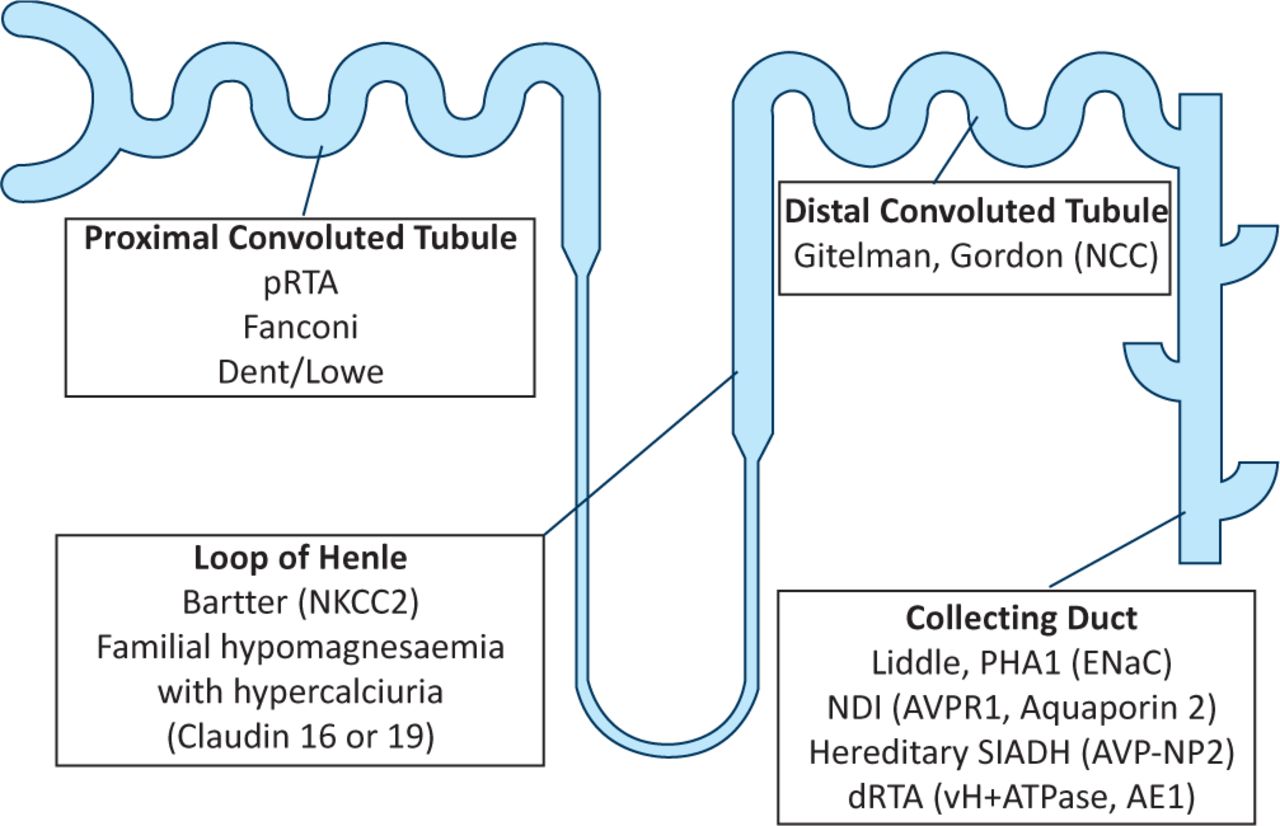
Renal tubular disorders.
The proximal tubule
The proximal convoluted tubule (PCT) is the main site of active transport and reabsorption of the majority of solutes present in the glomerular filtrate, as well as the location of the production of the key urinary buffer ammonium (NH4+):
In the early part of the PCT (S1), reabsorbed solutes include glucose, amino acids, phosphate, bicarbonate and various filtered low molecular weight proteins.
In the later part of the PCT (S2) urate is reabsorbed and secreted, and citrate is also reabsorbed.
Still further along is the proximal straight tubule (S3) where many drugs and their metabolites are secreted (eg loop and thiazide diuretics).
All this active transport depends on the ‘sodium pump’ (Na+K+–ATPase) on the basolateral side of the proximal tubular cell (PTC). This requires energy, so PTCs are full of mitochondria and are highly dependent on aerobic respiration, and are therefore vulnerable to hypoxia – one reason why PTCs are particularly susceptible to injury or necrosis from renal ischaemia and drug nephrotoxicity.
Disturbance of active transport processes
Failure of these active transport processes in the PTCs results in reduced reabsorption of the solutes already mentioned, which can then appear in the final urine.
Glucose. Various genetic defects affect glucose (isolated renal glycosuria) and amino acid (aminoacidurias) transport, such as cystine (dimeric cysteine) in cystinuria, one of the commonest clinically significant inherited defects of amino acid transport causing stones in humans. Cystinuria must be distinguished from cystinosis, a lysosomal storage disease affecting the PCT and due to the intracellular accumulation of cystine.
Bicarbonate. Disturbance of bicarbonate reabsorption presents as proximal renal tubular acidosis (pRTA or type 2 RTA). Initially, urine pH will be alkaline and systemic bicarbonate concentration will fall, causing an acidosis. When the threshold for bicarbonate reabsorption is exceeded (usually at a plasma or serum concentration of around 16 mmol/l) any bicarbonate not reabsorbed by the PCT is reabsorbed by the thick ascending limb of the loop of Henle and the collecting duct (together sometimes called the ‘distal nephron’). This limits further bicarbonate loss and the urine pH becomes more acid unlike distal RTA – see later. This form of RTA can occur in an isolated monogenic form1 and is also caused by carbonic anhydrase inhibitors (eg acetazolamide) or derivative drugs such as the anticonvulsant topiramate.
Renal Fanconi syndrome. However, pRTA is usually associated with uricosuria, glycosuria, phosphaturia, aminoaciduria and low molecular weight proteinuria, which comprises the renal Fanconi syndrome. This syndrome can occur in a number of acquired diseases such as myeloma2 and Wilson disease,3 but also as a side effect of some drugs (notably ifosfamide, tenofovir4 and aminoglycoside antibiotics) and in mitochondrial disease, which can be drug-related or inherited.
A genetic form of the renal Fanconi syndrome associated with nephrocalcinosis and nephrolithiasis occurs in Dent disease, a recessive X-linked condition due to a mutation in a PCT intracellular chloride transporter CLC-5,5 or in the intracellular phosphatase enzyme OCRL1, which causes a Dent-like syndrome known as Dent-2.6 OCRL1 mutations are also the cause of Lowe syndrome, an inherited renal Fanconi syndrome associated with mental retardation and congenital cataracts (oculocerebrorenal syndrome).
The loop of Henle
The loop of Henle is the site of the counter-current multiplier that serves to generate the corticomedullary osmotic gradient, and hence the kidney's ability to concentrate and dilute the final urine. This ability depends on the reabsorption of sodium (Na+) and chloride (Cl−) along the water impermeable thick ascending limb (TAL) of the loop of Henle (also known as the ‘diluting segment’).
Bartter syndrome
Bartter syndrome results from a failure of Na+ and Cl− reabsorption in the TAL, and thus a failure to concentrate the urine. This results in salt wasting, polyuria and volume depletion (often with hypotension). Consequent secondary hyperaldosteronism and increased delivery of Na+ to the downstream collecting duct lead to increased urinary excretion of potassium (K+) and hydrogen (H+) ions, producing hypokalaemia and metabolic alkalosis. The TAL is also the major site of calcium (Ca2+) and magnesium (Mg2+) reabsorption, which depends on normal NaCl reabsorption (see below). Thus, urinary losses of Ca2+ and Mg2+ are increased in Bartter syndrome and hypomagnesaemia is not uncommon in types 1 and 2 (see below).
NaCl reabsorption in the TAL relies on a number of transporters working in concert. The furosemide-sensitive apical transporter NKCC2 transports Na+, K+ and 2Cl− together into the cell, driven by the low intracellular Na+ concentration, maintained by the basolateral Na+–K+–ATPase. However, the K+ concentration in tubular fluid is much less than the concentrations of Na+ and Cl−. This would be limiting if it was not for an apical K+ channel, ROMK, which recycles K+ back across the apical membrane into the lumen. This K+ recycling generates a lumen positive potential difference that drives the reabsorption of Ca2+ and Mg2+ (and some Na+) paracellularly between the TAL cells. Meanwhile, Cl− is transported out of the TAL cell basolaterally via the Cl− channels, ClC-Kb and ClC-Ka, which are both regulated by an accessory protein known as barttin. Genetic mutations of any of these transport and regulatory proteins can cause Bartter syndrome by reducing NaCl transport along the TAL, with its local and downstream effects on Ca2+, Mg2+, K+ and H+ excretion.
Types of Bartter syndrome. Types 1–4 of Bartter syndrome are autosomal recessive.
Types 1 and 2 arise from NKCC2 and ROMK mutations, respectively, and can be associated with nephrocalcinosis.
Type 3 is caused by ClC-Kb mutations and has a milder phenotype, probably due to some redundancy in Cl− channel function.
A barttin mutation results in a more severe form known as type 4 which, because this protein is also present in the inner ear, is associated with sensorineural deafness.
Type 5 is an autosomal dominant form caused by activating mutations of the calcium sensing receptor (CaSR) on the basolateral membrane of TAL cells. CaSR activation inhibits NaCl reabsorption, explaining the renal effects of hypercalcaemia. Affected patients also have hypocalcaemia from parathyroid hormone suppression (due to parathyroid gland expression of the CaSR), nephrocalcinosis and stones (for review, see Ref. 7).
Familial hypomagnesaemia with hypercalciuria
Another inherited tubular disease affecting this nephron segment is familial hypomagnesaemia with hypercalciuria. This is due to a defect in the paracellular pathway (mentioned above) for Ca2+ and Mg2+ reabsorption, which depends on the selective permeability of cell junction proteins known as claudins. Mutations in claudin 16 or 19 cause this autosomal recessive syndrome with nephrocalcinosis and ill-understood recurrent urinary tract infections. It invariably progresses to renal failure.
The distal convoluted tubule
The distal convoluted tubule (DCT) is involved mainly in Na+ and Cl− transport, as well as some Ca2+ and Mg2+ reabsorption, although in this case across (transcellular) rather than between (paracellular) the DCT cells. Na+ and Cl− transport occurs via a thiazide-sensitive apical NaCl co-transporter (NCC).
Gitelman syndrome
Loss-of-function mutations of NCC cause Gitelman syndrome,8 characterised by:
milder renal salt losses and volume contraction than in Bartter syndrome
hypokalaemia and metabolic alkalosis, but with
hypocalciuria, similar to the effect of thiazide diuretic administration rather than the hypercalciuria of Bartter syndrome.
Gitelman syndrome is usually asymptomatic, often diagnosed late in childhood or incidentally in adulthood.
Gordon syndrome
The mirror image of Gitelman syndrome is the autosomal dominant (usually) Gordon syndrome (or pseudohypoaldosteronism type 2, or familial hyperkalaemic hypertension). This is caused by NCC overactivity due to mutations in the upstream regulators of NCC, WNK (With No lysine (K)) kinases 1 and 4.9 The result is hypertension, hyperkalaemia, metabolic acidosis and hypercalciuria. Patients with Gordon syndrome are particularly sensitive to thiazide diuretics which can correct most of its clinical features.
The collecting duct
The collecting duct comprises two main cell types:
Na+ and water reabsorbing, and K+ secreting, principal cells, and
acid or bicarbonate secreting intercalated cells.
Principal cells
Principal cells are the majority cell type and have the apical Na+ channel, ENaC and the K+ channel ROMK, as well as in the basolateral Na+–K+–ATPase, which is present in all polarised renal epithelial cells. The amiloride-sensitive ENaC is regulated by aldosterone, which increases both the number of open ENaCs and the activity of the Na+–K+–ATPase, and the number of ROMK channels. The net effect is to increase Na+ reabsorption and K+ secretion/excretion.
Liddle syndrome
Water reabsorption also occurs across the principal cells via an apical water channel, aquaporin 2, controlled by vasopressin (antidiuretic hormone). An autosomal dominant form of hypertension, Liddle syndrome, is caused by mutations in ENaC that prevent its removal from the apical membrane, thus maintaining increased ENaC activity.10 In addition to hypertension, there is hypokalaemia and metabolic alkalosis. Not surprisingly, patients with Liddle syndrome respond well to amiloride, whereas spironolactone is ineffective.
Apparent mineralocorticoid excess syndrome
A similar syndrome, apparent mineralocorticoid excess, is autosomal recessive and due to loss-of-function mutations in the enzyme 11-hydroxysteroid dehydrogenase-2 (11HSD2). The mineralocorticoid receptor (MR) in the collecting duct can bind cortisol (present in much higher concentrations than aldosterone), as well as aldosterone, and the intracellular enzyme 11HSD2 normally metabolises cortisol, preventing it from activating the MR in place of aldosterone. Licorice inhibits this enzyme, which can cause hypertension.
Pseudohypoaldosteronism type 1 (PHA1)
A mirror image of Liddle syndrome is pseudohypoaldosteronism type 1, which is like Addison's disease. Typical features are salt wasting, hypotension, acidosis and hyperkalaemia. The syndrome has two forms due to:
Autosomal recessive loss-of-function ENaC mutations unresponsive to aldosterone (type 1a). There is wide distribution of ENaC in the lung, kidney, skin and gastrointestinal tract, so the phenotype is often more severe.
Autosomal dominant mutations of the MR that cannot bind aldosterone (type 1b). In this form the phenotype is milder and see is confined to the kidney.10
Hereditary nephrogenic diabetes
Hereditary nephrogenic diabetes insipidus (NDI) is a condition of resistance to the action of vasopressin caused either by:
recessive loss-of-function mutations in the V2 vasopressin receptor gene (AVRP2) on the X chromosome (>90%),11 or
recessive and autosomal dominant mutations in the water channel aquaporin 2 gene (AQP2).12
Vasopressin levels are elevated and the main features are polyuria, nocturia and polydipsia, usually associated with mild hypernatraemia. The urine concentrating ability is lost, so urine osmolality is low and plasma or serum osmolality raised. Thirst is normal, so severe hypernatraemia is uncommon. Lithium interferes with vasopressin signalling via cyclic AMP and can cause an acquired form of NDI (a side effect of its chronic use) because it enters the principal cells through ENaC. This effect can be blocked or ameliorated by amiloride.13
Hereditary central diabetes insipidus
Hereditary central diabetes insipidus is due to autosomal dominant mutations in the AVP-neurophysin II gene (AVP-NPII), leading to loss of vasopressin secretion (unlike NDI, this is not usually evident at birth, but progressive) and circulating vasopressin levels are low.
Hereditary syndrome of antidiuretic hormone secretion (SIADH) and hyponatraemia
In contrast to X-linked NDI (see above) SIADH can be due to a gain-of-function mutation in the AVRP2 gene and V2 vasopressin receptor.14 A loss-of-function polymorphism of the TRPV4 cation channel has also been linked to SIADH and hyponatraemia, and it seems that this ion channel is involved in osmosensing by the hypothalamus.15 Those affected have a blunted response to hypotonicity and behave as if they have a reset osmostat, and can regulate their plasma osmolality normally, although at a lower than normal value. This polymorphism has a dominant-negative effect on the normal wild type allele.
Alpha intercalated cells
Alpha intercalated cells excrete acid into the urine by generating H+ from the intracellular conversion of CO2 and water to carbonic acid, followed by its breakdown to bicarbonate and H+ catalysed by carbonic anhydrase 2 (CA2). The H+ is secreted into the tubular lumen by the electrogenic H+ vH-ATPase and the bicarbonate transferred to blood via the Cl−/bicarbonate anion exchanger AE1 (SLC4A1).
Hereditary distal renal tubular acidosis
Hereditary distal renal tubular acidosis (type 1 RTA or dRTA) can be caused by loss-of-function mutations of the subunits B1 or a4 of the vH-ATPase, or of AE1. An impaired ability to excrete acid in the urine can lead to metabolic acidosis, complicated by rickets, osteomalacia or reduced bone mineral density, nephrocalcinosis and stones. There is also increased urinary potassium excretion, leading to hypokalaemia, although this is more difficult to explain. The tendency to form calcium phosphate stones is because of the alkaline urine and hypercalciuria in acidotic patients (so-called ‘complete’ dRTA); urine pH is always above 5.3 in the presence of a systemic acidosis. Patients can still have an acidification defect but without acidosis (‘incomplete’ dRTA). If this is suspected (in the presence of nephrocalcinosis or with a family history of stones), a urinary acidification test is necessary.16,17
Autosomal recessive mutations of the B1 subunit of the vH-ATPase (also present in the inner ear) cause dRTA with congenital sensorineural deafness.18 With autosomal recessive a4 mutations, the onset of deafness is often later in early adulthood. AE1 mutations cause autosomal dominant dRTA without deafness, which can present in childhood with rickets or in later life with recurrent renal stones and nephrocalcinosis. This form of dRTA is often recessive in the tropics due to associated inherited red cell defects, such as South-East Asian ovalocytosis.19 CA2 mutations can cause a rare mixed type of RTA with both pRTA and dRTA features, associated with osteopetrosis and cerebral calcification.20 Acquired dRTA is more common in adult clinical practice and typically is associated with autoimmune diseases, especially Sjögren syndrome.21
Conclusions
This article is not an exhaustive account of renal tubular disorders but has covered many of those likely to be encountered clinically, especially in adult patients. The intention has been to link structure with function, and to make it easier to remember and understand the pathophysiology of the tubulopathies described.
- © 2012 Royal College of Physicians








{kind=link}