
Introduction
Tay-Sachs disease was one of the first lysosomal diseases to be described as a clinical entity.1,2 Originally classified as a cause of ‘familial amaurotic idiocy’, this devastating neurodegenerative disorder was first noted in Jewish infants3 – most of whom were the offspring of parents settled in the newly ghettoised cities of Western Europe and the USA, who had escaped persecution in Russia and neighbouring countries. Tay-Sachs disease is one of three GM2 gangliosidoses, a group of disorders with extensive involvement of the neurological system.1–3 They represent an extremity of neurodegeneration that challenges the societal provision of resources and for which major scientific investment will be needed to introduce any definitive therapy. Tay-Sachs and Sandhoff diseases are ultra-orphan disorders that illustrate several critical principles in the burgeoning field of human metabolism:
the unusual ethnic distribution of many hereditary disorders
the challenge of managing a relentless neurodegenerative disease that presents in infancy in the context of modern developed medicine
striking clinical and genetic heterogeneity, which reflects the intimidating biochemistry of sphingolipid metabolism, the role of the three mammalian β-hexosaminidase isozymes and interactions with an activating factor (GM2 activator protein)
ethical principles for the application of biochemical and genetic screening in at-risk populations
discoveries in molecular cell biology and the use of naturally occurring and transgenic models to explore pathogeneses, as well as innovative therapeutics of the human brain.
History
Waren Tay was a surgeon who practised in the hospitals of London's East End; in the late 19th century he was based principally at The London Hospital, Whitechapel. Although a generalist, he nonetheless followed his own particular specialty – ophthalmology – and described in the very first volume of the Transactions of the Ophthalmological Society (1881) ‘symmetrical changes in the region of the yellow-spot in each eye in an infant’.2 Tay's paper shows the extraordinary ‘cherry red’ spot in the region of the macula, which contrasted strongly with a surrounding white patch. Tay followed up the female infant and noted that she became passive and weak after six months with arrested cerebral development and atrophic optic discs. Subsequently, Tay reported two further children from the same family and another affected child. None of his great contemporaries (including Robert Hutchinson and Hughlings Jackson) were able to contribute insights into the nature of the condition. Bernard Sachs, one of the founding fathers of American neurology, reported his first case in 1887, including careful necropsy findings.3 Dr Sachs subsequently reported many patients in the Ashkenazi Jewish population of Brooklyn, New York, continuing to study the disease as he moved to Mount Sinai Hospital. Sachs reported that practically all neurones in the brain were abnormal, with pathological displacement of the nucleus by accumulated material in the cytoplasm. Later investigators showed axonal and white-matter abnormalities, with formation of atypical dendrites and ectopic neuritogenesis. Histochemical stains later revealed lipid storage, and ultrastructural studies showed abnormal lysosomes in neurones, often with a laminar distribution of storage material described as ‘membranous cytoid bodies’.
Tay-Sachs disease, a devastating neurological condition with death in the first few years of life, is no longer seen as a disease restricted to the Ashkenazi Jewish population; moreover, it occurs in infants, young children and adults of all ages. Inherited as an autosomal recessive condition, with a high carrier frequency in the Ashkenazi Jewish population, like the related GM2 gangliosidoses Sandhoff disease,4 the condition is pan-ethnic.1 Late-onset forms of GM2 gangliosidosis may come to light in adults with unusual or typical neurological complaints, ranging from autonomic neuropathy to lone cerebellar ataxia; this disease however, most often mimics amyotrophic lateral sclerosis (motor neurone disease).
Inborn errors of lysosomal metabolism
Inborn errors that affect activity of lysosomal hydrolases are susceptible to functional correction by harnessing molecular recognition signals, such as mannose-6-phosphate, for which receptors exist on the lysosomal membrane and the plasmalemma. Dr Elizabeth Neufeld and colleagues demonstrated mutual correction of the biochemical defect in cultured fibroblasts from the genetically distinct Hurler (mucopolysaccharidosis (MPS) 1) and Hunter (MPS2) diseases as a result of secretion of corrective factors into the culture medium.5 These factors are phosphorylated forms of the enzymes that were reciprocally deficient in each disease. This secretion-recapture system paved the way for enzymatic augmentation, and eight enzyme preparations for lysosomal diseases are now licensed by biopharmaceutical companies (Box 1). An alternative delivery system, based on the mannose-lectin displayed on the surface of the target cells (macrophages) in Gaucher disease, has had an unprecedented therapeutic success and has stimulated massive investment. Administration of therapeutic proteins that display appropriate recognition markers for cellular uptake and delivery to the lysosomal compartment has proved very attractive for development of orphan medicinal products.6
Lysosomal diseases for which treatments are licensed.

Biochemistry and genetics
GM2 gangliosidoses are sphingolipidoses in which there is accumulation of undigested glycosphingolipids, of which GM2 ganglioside and its asialo-derivative, the sphingolipid GA2, are members.1,4 Other lysosomal diseases include the mucopolysaccharidoses, glycoproteinoses and other miscellaneous disorders, such as those affecting lysosomal biogenesis and lymphocyte activation (Table 1).
Inborn errors of lysosymal metabolism.
The structure of GM2 ganglioside was unequivocally identified only in 1963. The term GM2 gangliosidosis applies to Tay-Sachs disease and the related Sandhoff disease – a condition that is not over-represented in the Jewish population but occurs in numerous other communities, especially in certain isolates.4 Tay-Sachs disease is caused by deficiency of the acidic, heat-labile enzyme β-hexosaminidase A. In Sandhoff disease, the isozyme β-hexosaminidase B is also deficient.4 β-hexosaminidase A cleaves an N-acetylgalactosamine residue from the branched-chain glycan terminal structure of naturally occurring GM2 ganglioside in a reaction that requires the GM2 activator (a genetically distinct sphingolipid-binding protein). β-hexosaminidases A and B are lysosomal hydrolases, which are heterodimers and homodimers of the cognate α- and β-subunit proteins, respectively: Tay-Sachs disease is caused by mutations in the HEXA gene that encodes the β-subunit, while mutations in the HEXB gene affect the β-subunit common to the A and B isozymes and cause Sandhoff disease.1 The crystal structures of hexosaminidases A and B have been solved.
Carrier screening
On account of the high risk of Tay-Sachs disease in the Ashkenazi Jewish population (carrier frequency one in 27, homozygotes potentially in one in 2,900 live births), there has been a need to identify heterozygous carriers and to study at-risk couples and monitor pregnancies. An international carrier screening programme that also allows for prenatal diagnosis in those requesting it was pioneered by Dr Michael Kaback.1,7 About 1.5 million people have been screened using enzymatic methods with plasma followed by genetic confirmation, which has greatly reduced the burden of Tay-Sachs disease in the Ashkenazi Jewish population. The frequency of heterozygotes is about one in 300 of the European and American gentile population but is increased in particular groups, including Irish Americans (one in 50) and French Canadians and Cajun people, in whom the carrier frequency is one in 30.
Genotype-phenotype correlations
The genetic and clinical heterogeneity reflects, in part, the involvement of several proteins in the same lysosomal reaction (the cleavage of the neutral N-acetylgalactosamine from GM2 ganglioside). β-hexosaminidase B also catalyses hydrolysis of glycosaminoglycans, so patients with Sandhoff disease may have biochemical features of the mucopolysaccharidoses, with increased urinary glycosaminoglycans and storage of glyco protein and glycosaminoglycans in systemic tissues, occasionally in association with hepatomegaly and minor skeletal dysmorphism. Several proteins thus participate in distinct biochemical reactions, and disease-related mutations will have disparate effects on lysosomal breakdown of glycoconjugate substrates. Molecular analysis of the HEXA and HEXB genes has revealed numerous mutations responsible for GM2 gangliosidosis in all populations as part of an ever-diversifying phenotype-genotype spectrum.1
Biogenesis of soluble proteins of the lysosomal matrix
Many lysosomal proteins undergo cotranslational processing in the endoplasmic reticulum and Golgi apparatus, and the nascent hexosaminidase subunit proteins are no exception. These polypeptides acquire the appropriate phosphorylated molecular recognition signal (mannose-6-phosphate) that ensures delivery to their site of action and destination in the lysosome itself.
Treatment of lysosmal diseases
In general, the pharmacopoeia for rare diseases is dismal: until the introduction of orphan drug legislation, first in the USA and subsequently in Europe, the development costs for drugs in rare disorders prevented investment. Early realisation by the discoverer of the lysosome, Christian De Duve, that the organelle has access to the external fluid phase gave the impetus to therapeutic endeavour. Recombinant therapeutic proteins decorated by the appropriate recognition signals for molecular targeting to the lysosome have long been studied. Inspired by the spectacular success of alglucerase (tissue-derived, macrophage-targeted glucocerebrosidase) and imiglucerase (its recombinant successor) for Gaucher disease, considerable investment in this field has been made by biotechnology and pharmaceutical companies 6 (see Box 1).
Molecular targeting to the brain
Most patients with Tay-Sachs disease are infants who die within the first years of life, with an unremitting and relentless condition affecting the entire central nervous system.8,9 The use of inhibitors of substrate formation (substrate deprivation therapy) or the experimental use of pharmacological chaperones,10 which would stabilise unstable nascent proteins and prevent their premature destruction before arrival at their site of action in the lysosome, are theoretical possibilities for treatment.6 More radical therapy is needed to correct the profound defect of ganglioside metabolism in neural cells. The blood-brain barrier is also an obstacle to the passage of recombinant enzymes and, although recent attempts have been made to improve lysosomal diseases by the use of either systemic or intrathecal and intraventricular administration of corrective proteins, with formidable difficulties for long-term administration, the failure of several clinical trials gives scant encouragement that this stratagem will have long-term application.
Gene therapy
The existence of the lysosomal secretion-recapture system offers an attractive basis for a radical approach for using gene therapy vectors, which will allow the transfer of therapeutic genes to particular sites in the brain and spinal cord, from where the encoded active protein can be distributed widely.5 Examples already exist in which gene therapy has been successful in whole-animal models of lysosomal diseases affecting the brain. Latterly, lentiviral vectors have been used to transduce haematopoietic stem cells, which serve as a source of healthy microglia and also act as Trojan horses to deliver the therapeutic product after transmigration across cerebral capillaries.
Direct use of non-pathogenic adeno-associated viral vectors, which remain episomal but harness the protein-synthesising machinery of neural cells to express therapeutic proteins, have recently been developed. The expressed enzymes seem to be appropriately processed for local diffusion, with dissemination and functional complementation throughout the brain. Given at the appropriate time, this intervention has shown remarkable effects on neurological function and survival in challenging acute neurodegenerative models11–12 (see Fig 1 and Ref 11 and accompanying data).
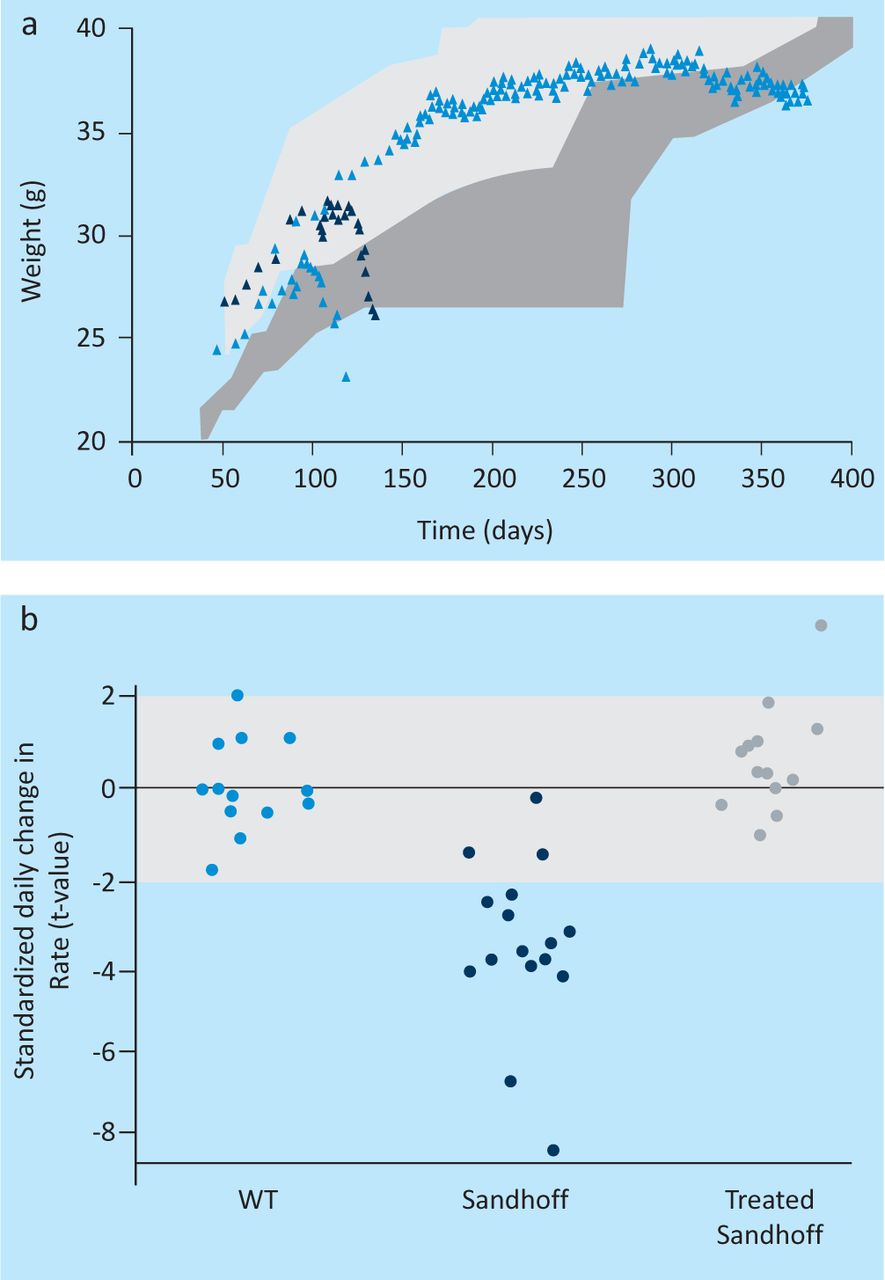
Successful gene therapy in mice with GM2 gangliosidosis using recombinant adeno-associated viral vectors (rAAV). (a) Survival, weight and neurological function after gene transfer of human β-hexosaminidases A and B using rAAV vectors in HEXB knockout (Sandhoff) mice with acute GM2 gangliosidosis. (b) Weight and rescue of neurological function assessed in wildtype (WT), untransduced (Sandhoff) and transduced Sandhoff (treated Sandhoff) mice. Transduced animals were injected with rAAV at the age of four weeks. See also reference 11 (free access, with accompanying videos).
Summary
This short paper has reviewed progress in the development of lysosomal therapeutics, with particular reference to disorders that affect the brain and for which spectacular results in experimental systems provide grounds for optimism.11,12 Future development will be predicated on experimental medicine as revealed by challenging safety and efficacy trials as a critical part of the orthodox clinical testing pathway now dignified by the term translational medical research.
- © 2012 Royal College of Physicians








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.