
Introduction
In Mendelian disorders, there is a direct causal relation between mutation and disease phenotype. If it is possible to understand what effects the underlying mutation has on cell and organ function, then it might also be possible to use a molecularly targeted approach to treat the disease.1 Tuberous sclerosis is one of the first Mendelian disorders for which the identification of causative genes and their subsequent functional characterisation has led to clinical trials of molecularly targeted agents. This condition is caused by mutations in either of two genes, TSC1 or TSC2, and is characterised by the growth of tumours in multiple organs, seizures and intellectual disability. Targeted therapies for a range of the manifestations of the disease are being developed.
The genetics of tuberous sclerosis
TSC1 is located on chromosome 9 and TSC2 on chromosome 16. The genes encode two proteins, TSC1 and TSC2, which form a complex within cells. Over 50 proteins interact with the TSC1/TSC2 complex; however, the best-characterised function of the complex is the regulation of mammalian target of rapamycin complex 1 (mTORC1), a protein kinase that controls many cellular functions (Fig 1).2
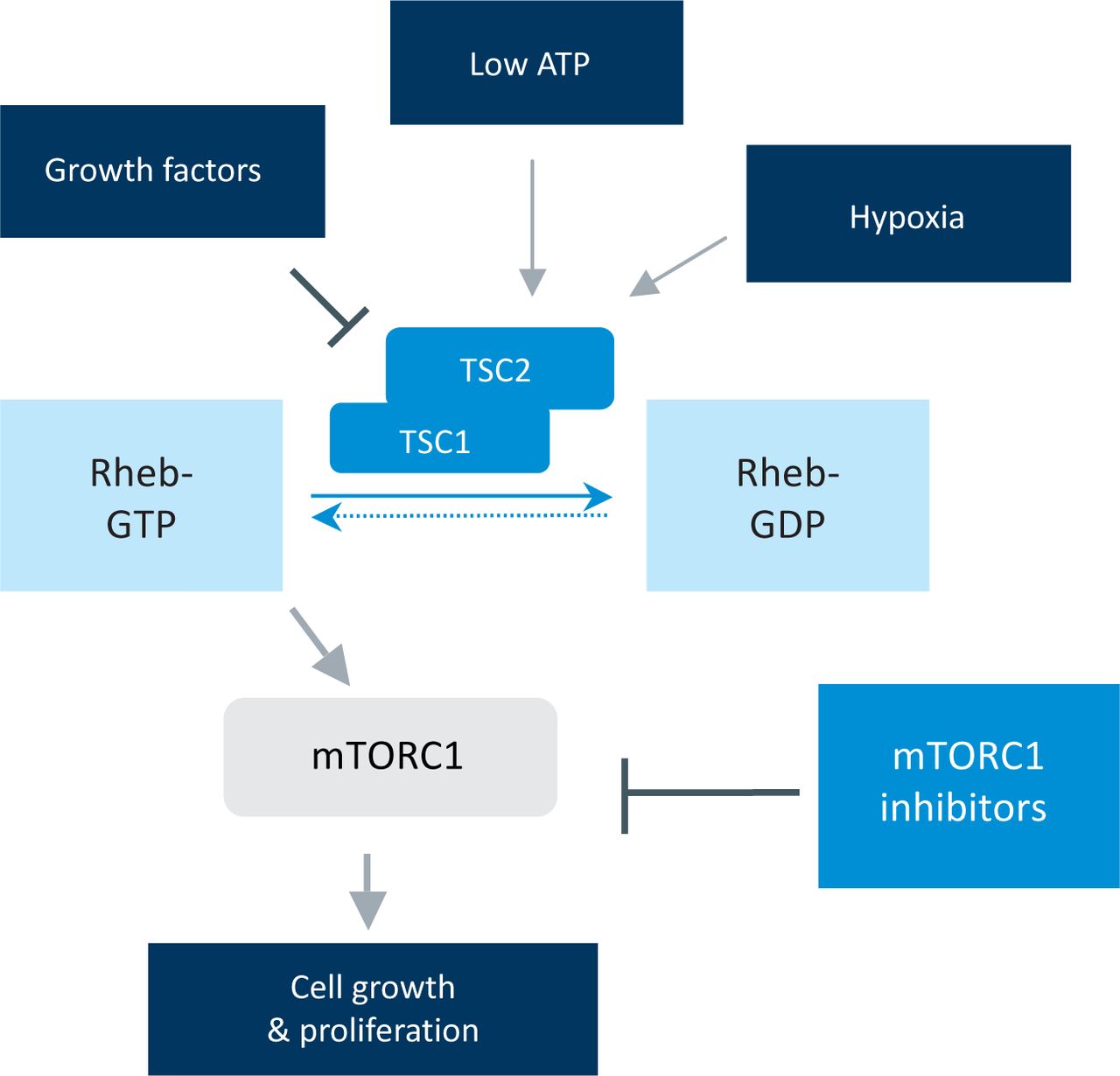
Signalling inputs into the mTORC1 pathway. The TSC1/2 genes integrate multiple stimuli into the control of the mTORC1 kinase. mTORC1 = mammalian target of rapamycin complex 1; Rheb = Ras homolog enriched in brain; TSC1/2 = tuberous sclerosis 1/2.
TSC2 is a GTPase-activating protein (GAP) that converts a G-protein, Ras homolog enriched in brain (Rheb), from an active GTP-bound form to an inactive GDP-bound form. The active form of Rheb in turn activates mTORC1. Many signalling pathways converge on the TSC1/2 complex. Growth factor stimulation leads to an inhibition of TSC2 GAP activity, whereas hypoxia or low intracellular energy levels activate TSC2.
Although tuberous sclerosis is an autosomal dominant condition, approximately two-thirds of cases result from a new mutation, with neither parent being affected. Pathogenic mutation in TSC1 or TSC2 leads to loss of TSC2 GAP activity, maintenance of Rheb in its active form and activation of mTORC1. In addition to a germline TSC1 or TSC2 mutation, an acquired ‘second hit’ mutation in the other TSC1 or TSC2 allele is also required for the formation of most lesions in tuberous sclerosis, consistent with the two-hit tumour suppressor model of Knudson. However, some of the manifestations of tuberous sclerosis, such as cognitive impairment, might reflect the effects of haploinsufficiency, a gene-dosage effect where loss of only one allele is sufficient to impair cellular functioning.
The tumours that occur in tuberous sclerosis are relatively molecularly homogeneous in the sense that they carry mutations in either TSC1 or TSC2 and exhibit mTORC1 activation (Fig 2). This relative molecular homogeneity means that an effective molecularly targeted agent is likely to work in most patients. Also, as mTORC1 activation underlies many of the manifestation of this multisystem disorder, mTORC1 inhibitors might be beneficial for a range of these manifestations.

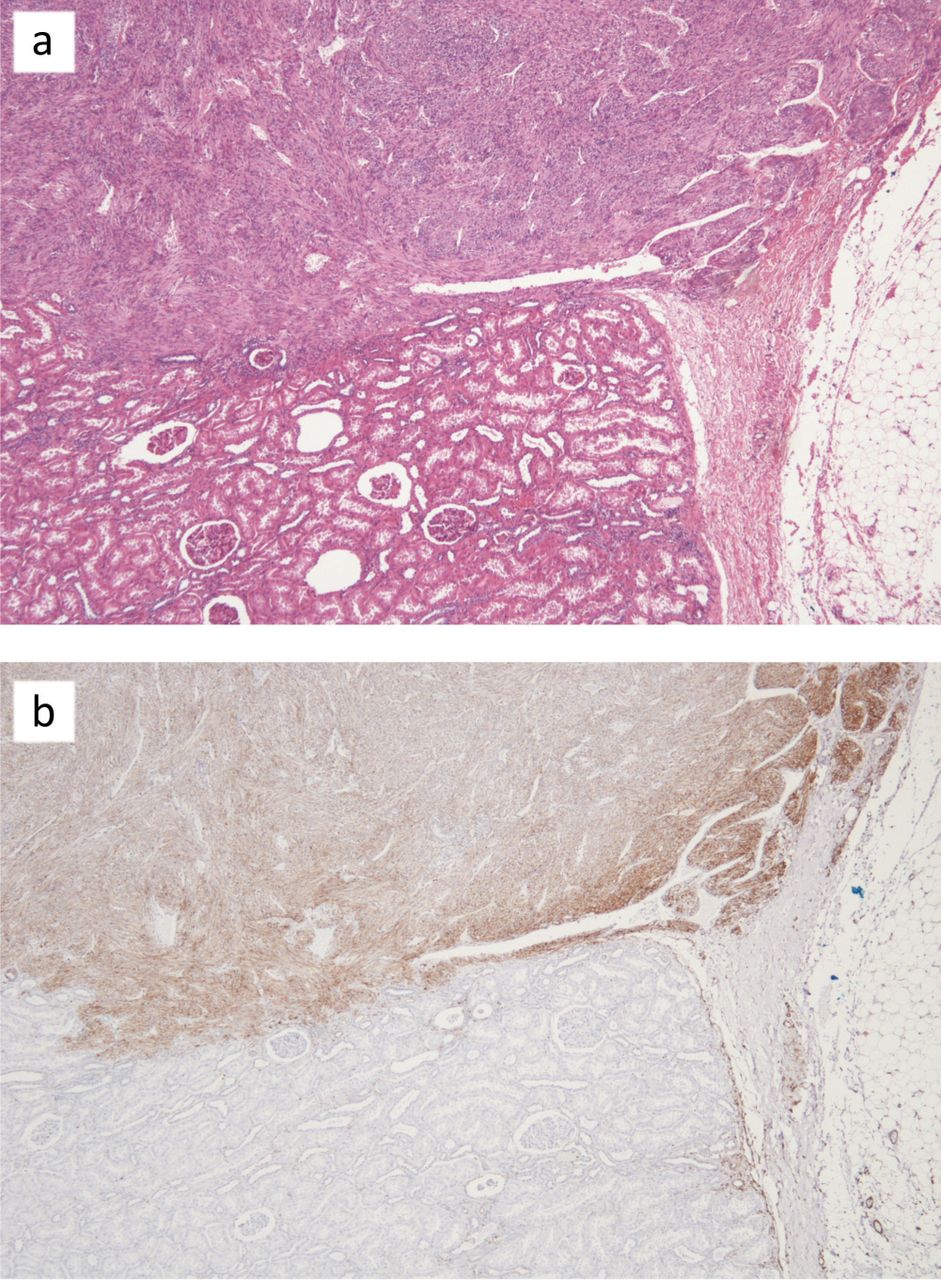
mTORC1 pathway activation in a renal AML. (a) shows a renal AML adjacent to normal kidney tissue. (b) shows a similar section immunohistochemically stained with an anti-phospho S6 antibody, which is marker of mTORC1 activation. AML = angiomyolipoma; mTORC1 = mammalian target of rapamycin complex 1.
The clinical features of tuberous sclerosis
Tuberous sclerosis occurs in approximately 1 in 10,000 people. It affects many organs and has a variable clinical phenotype,3 with many features having an age-related penetrance. The multisystem nature of the disease means that patients with tuberous sclerosis can have complicated care needs and coordination of this care is difficult.
Neurological
Most patients with tuberous sclerosis have epilepsy and this is often the presenting feature of the condition. Although complex partial seizures are most commonly observed, a variety of seizure types can occur. Infantile spasms occur in one-third of infants with tuberous sclerosis and are typically characterised by clustered repetitive flexion of the limbs. However, repetitive extension of the limbs, a mixture of flexion and extension or more subtle signs, such as head nods, upward eye deviation or shoulder elevation, can occur. Onset peaks at 4–6 months of age. There is a characteristic chaotic and high voltage interictal electroencephalography (EEG) pattern which, when typical, is termed ‘hypsarrhythmia’. Infantile spasms are associated with severe developmental delay and cognitive impairment, although this can be ameliorated by prompt diagnosis and treatment.
There is a great variation in the global intellectual ability of people with tuberous sclerosis. Most fall within a normal range, but approximately one-third are profoundly impaired. Even the most able of individuals with tuberous sclerosis often have specific neurocognitive deficits, such as in executive control processes, attentional skills and memory skills. Developmental disorders, such as autism, autistic spectrum disorder and attention deficit hyperactivity disorder, are commonly associated with tuberous sclerosis, as are mood and anxiety disorders. At the behavioural level, problems such as aggression, impulsivity and sleep difficulties are also common.
Several macroscopic brain lesions occur in people with tuberous sclerosis. Approximately 80–90% of patients have cortical tubers, which are focal developmental abnormalities of the cerebral cortex, and/or subependymal nodules (SENs). These are asymptomatic nodules found on the walls of the lateral ventricles. Subependymal giant cell astrocytomas (SEGAs) are low-grade glioneuronal tumours that develop in approximately 5–20% of patients. SEGAs usually develop adjacent to the foramen of Monroe and can cause hydrocephalus.
Renal
Renal angiomyolipomas (AMLs) occur in up to 80% of adults with tuberous sclerosis, in whom they are typically multiple and bilateral. Rupture of blood vessels in AMLs can cause life-threatening haemorrhage. Renal cysts are common in patients with tuberous sclerosis and are usually asymptomatic; however, a polycystic kidney phenotype occurs in those patients with a contiguous TSC2 and polycystin-1 (PKD1) deletion, who often progress to renal failure in early adult life.
Pulmonary
Lymphangioleiomyomatosis (LAM) is a disorder of the lungs and lymphatics that can occur sporadically or in association with tuberous sclerosis.4 The sporadic form is caused by acquired biallelic mutations in TSC2. LAM is a rare condition, occurring almost exclusively in women between menarche and menopause, with a prevalence of approximately 5 in 1,000,000 in the whole population. However, it is more common in patients with tuberous sclerosis. Radiological evidence of LAM is present in 40% of adult females with tuberous sclerosis, although only 2–3% will develop symptoms. The clinical features result from the accumulation of ‘LAM’ cells in the pulmonary interstitium and the lymphatic system. Respiratory symptoms tend to dominate the clinical course, with the most common problems being progressive dyspnoea and pneumothorax. Cystic destruction of the lung can lead to respiratory failure. Current evidence suggests that LAM can be considered a low-grade metastatic neoplasm. LAM cells arise from an unknown source, possibly AMLs, and migrate to the lung. Renal AMLs are found in approximately half of patients with sporadic LAM.
Skin
Tuberous sclerosis-associated skin lesions include hypomelanotic macules, ‘confetti’ macules, facial angiofibromas, forehead plaques, shagreen patches and ungual fibromas. Hypomelanotic macules are seen in most patients with tuberous sclerosis. They are present at birth or arise during early infancy. Some patients have numerous small, hypopigmented macules termed confetti lesions. Facial angiofibromas generally appear at approximately 2–5 years of age and eventually affect 75–90% of patients. They are red/reddish-brown papules found on the central face, especially in the nasolabial folds. Forehead plaques are found in 20–40% of patients with tuberous sclerosis and are present at birth or develop before the age of 10 years. Shagreen patches are irregular plaques that most commonly arise in the lower lumbar area in approximately 50% of patients. They can be present at birth, but most commonly arise during the first 10 years of life. Ungual fibromas are nodules arising under the proximal nail fold or under the nail plate. They also usually develop after the first decade.
Cardiac
Cardiac rhabdomyomas appear in utero at 22–28 weeks of gestation and are often detected by antenatal scans, where they are associated with a substantial risk of tuberous sclerosis, especially if the lesions are multiple. They usually regress during childhood and are normally asymptomatic, but can be associated with obstructive heart failure or arrhythmias.
Diagnostic criteria for tuberous sclerosis
A diagnostic triad for tuberous sclerosis of epilepsy, intellectual disability and facial angiofibroma (formerly know as adenoma sebaceum) was proposed during the early 20th century by Voght and Campbell. However, with today's understanding of the disease, this triad lacks sensitivity. The current consensus diagnostic criteria are shown in Table 1. The criteria are divided into ‘major’ features, which are considered to be more specific for tuberous sclerosis, and ‘minor criteria’ whose specificity is lower. A diagnosis of tuberous sclerosis is considered definite if two major features or one major feature plus two minor features are present, probable if an individual has one major feature plus one minor feature, and possible if one major feature or two or more minor features occur. Because of their lack of specificity, neither intellectual disability nor epilepsy form part of the diagnostic criteria; neither are the results of genetic testing included. However, genetic testing can be useful, particularly in children, who are too young to manifest enough features to fulfil diagnostic criteria.
Diagnostic criteria for tuberous sclerosis.12
Molecularly targeted therapy in tuberous sclerosis
Manipulation of mTORC1 signalling might be beneficial for wide range of features of tuberous sclerosis.5 Use of mTORC1 inhibitors in cell culture and murine models of tuberous sclerosis is associated with normalisation of mTORC1 signalling and regression of lesions. Several mTORC1 inhibitors are available for clinical use for other indications and could potentially be repositioned for the treatment of tuberous sclerosis. For example, sirolimus (also known as rapamycin) is licensed for use as an immunosuppressant in organ transplantation and everolimus for use in renal cancer and lymphoma.
In October 2010, the US Food and Drug Administration (FDA) granted accelerated approval for the use of everolimus to treat patients with SEGA associated with tuberous sclerosis who required therapy but who were not candidates for surgical resection. In September 2011, everolimus was approved in the European Union for the same indication. The efficacy and safety of SEGA was demonstrated in a single-arm open-label clinical trial.6 In the trial, 28 patients whose SEGA lesions had demonstrated growth on serial imaging received everolimus orally once daily at a starting dose of 3 mg/m2/d, with subsequent titration to serum trough level of 5–15 ng/ml. The median age of enrolled patients was 11 years (range 3–34 years). The primary efficacy endpoint was a reduction in the volume of their largest SEGA volume at six months. In total, 21 patients (75%) experienced a reduction of at least 30% and nine (32%) experienced a reduction of at least 50%.
The effect of sirolimus on lung function in patients with LAM has been investigated in a randomised, placebo-controlled study.7 Eligibility criteria included a diagnosis of LAM (either tuberous sclerosis associated or sporadic) and a forced expiratory volume in 1 sec (FEV1) after bronchodilatation of 70% of the predicted value or less. The primary outcome measure was assessed as the rate of change in FEV1 per month. In total, 89 patients were randomised, eight of whom had tuberous sclerosis. In the sirolimus group, most patients were maintained at a sirolimus trough level of 5–15 ng/ml. During a 12-month treatment period, the FEV1 slope was −12 +/−2 ml/month in the placebo group and 1 +/− 2 ml in the sirolimus group. These results indicate that sirolimus might have a useful role in the treatment of patients with moderately severe LAM.
The use of mTORC1 inhibitors to treat tuberous sclerosis-associated renal AMLs has been studied in several early phase clinical trials. The Trial of Rapamycin Therapy for Patients with Tuberous Sclerosis Complex and Sporadic LAM was a single-centre, single-arm phase I/II trial conducted in 25 patients, 19 of whom had tuberous sclerosis, six of whom had sporadic LAM, and all of whom had AMLs.8 Patients were treated for 12 months with dose escalations up to 15 ng/ml and were followed for an additional 12 months off therapy. In total, 20 patients completed 12 months of therapy and 18 completed 24 months of follow up. After 12 months of therapy, mean AML volume had decreased to 53% of baseline; however, there was tumour regrowth to 86% of baseline during the subsequent year of follow up off therapy.
The multicentre phase II non-randomised Trial of Efficacy and Safety of Sirolimus for Angiomyolopma in Tuberous Sclerosis and Sporadic LAM (TESSTAL) recruited 16 patients (10 of whom had tuberous sclerosis and six of whom had sporadic LAM)9. The participants received oral sirolimus for up to two years at trough levels in the range of 3–10 ng/ml. The response rate, as defined using the Response Evaluation Criteria in Solid Tumours (RECIST), was 50%. Most shrinkage occurred in the first year of treatment, but was maintained during the second year of treatment.
Dabora et al conducted a multicentre phase II trial of sirolimus for tuberous sclerosis-associated AMLs, where the primary outcome measure was the response rate of AMLs as defined using RECIST.10 In total, 36 patients were enrolled (although only 28 were evaluable at 12 months) and were treated with daily sirolimus maintained at a targeted concentration of 3 nm/ml for the first 15 weeks, which was subsequently escalated up to 15 mg/ml if there was no evidence of a response. After 52 weeks of treatment, the overall response rate was 16/36 (44.4%) and all participants were partial responders.
Drug-related adverse events were common in these trials, although most were low grade and self limiting. Commonly occurring adverse events included oral mucositis, acneiform rash, upper respiratory tract infections, nausea, diarrhoea, hyperlipidaemia and proteinuria. It appears that the adverse effect profile seen in patients with tuberous sclerosis in these trials is similar to that in other groups taking mTORC1 inhibitors, such as renal transplant patients.
It has been hypothesised that a major contribution to seizures and intellectual disability in patients with tuberous sclerosis is abnormal cellular functioning resulting, in turn, from abnormal regulation of mTORC1.11 Studies in mouse models of tuberous sclerosis suggest that use of mTORC1 inhibitors could lead to improved seizure control and cognitive function. Randomised controlled trials are currently being set up or are underway to investigate whether these improvements in mice also occur in humans.
The next steps
There is no doubt that the use of mTORC1 inhibitors alters the natural history of tuberous sclerosis. However, questions remain about which patients with tuberous sclerosis to treat, and when to start and how long to continue treatment. There might be a role in using mTORC1 inhibitors to prevent some manifestations of tuberous sclerosis, with therapy perhaps beginning in early childhood. However, the long-term safety of mTORC1 inhibition in this patient group needs to be studied further. Larger scale randomised, placebo-controlled trials to assess the effect of everolimus on SEGAs and AMLs are close to reporting and will provide further information to guide the management of patients with tuberous sclerosis.
- © 2012 Royal College of Physicians
References








{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.