
Key points
Mitochondrial DNA disorders are a common cause of inherited disease, affecting 1 in 5,000 of the UK population1
They should be considered in any complex multisystem disorder, especially those disorders in which neurological, ocular or endocrine features predominate
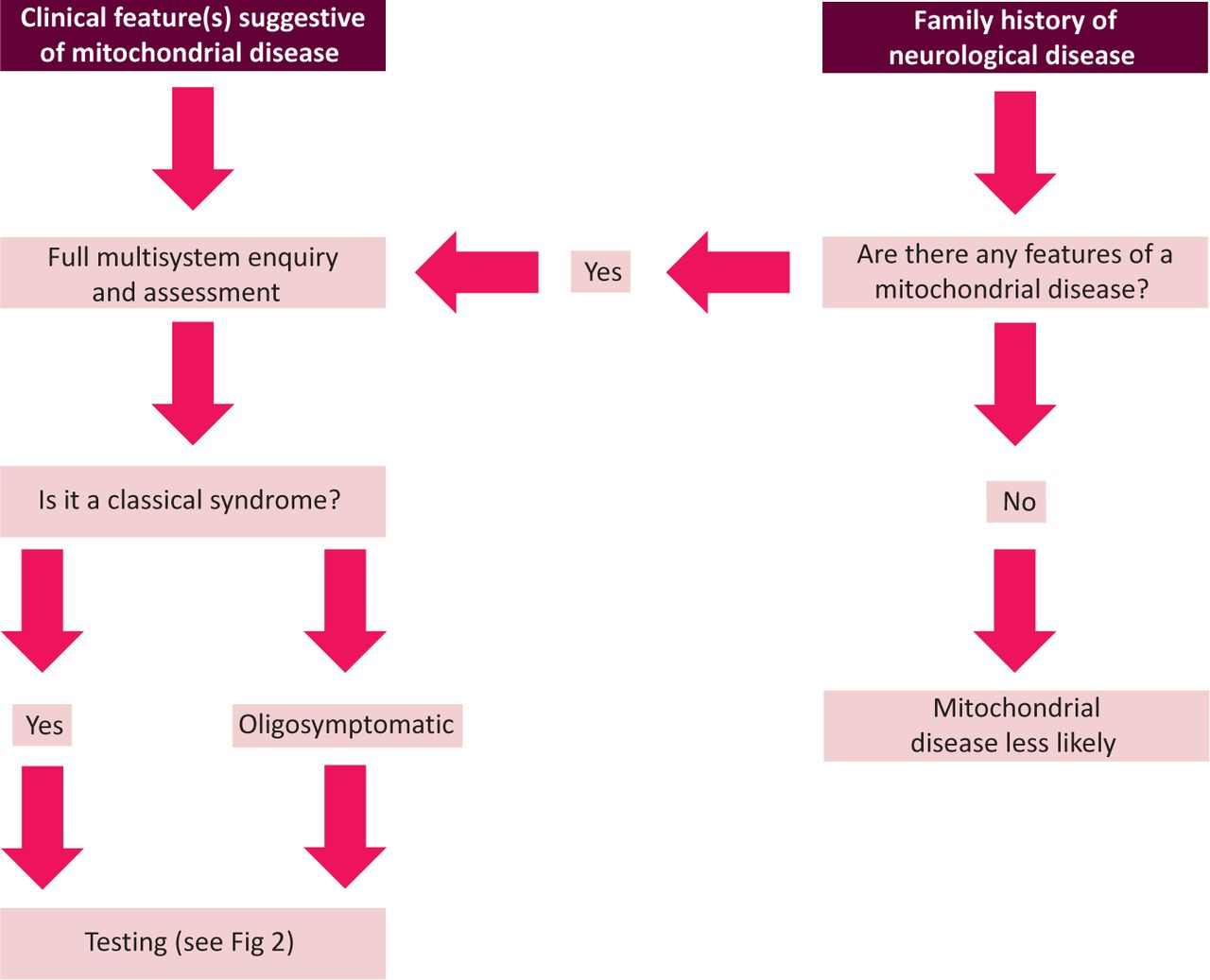
Many patients will fit neatly into defined classic syndromes, but significant numbers are oligosymptomatic, or have an overlapping, poorly defined, phenotype
If a mitochondrial disorder is suspected, it is vital to assess fully the clinical phenotype in the patient and their relatives with symptoms, paying particular attention to the non-neurological manifestations, because these can often be effectively managed
Counselling depends on the underlying molecular basis of disease and might necessitate discussion with specialist centres
There are currently no treatments known to modify the underlying disease process
Investigation of mitochondrial disorders can be complex and might necessitate referral to one of the three commissioned groups forming the Rare Mitochondrial Disorders Service for Adults and Children (in Newcastle, Oxford and London)
Numerous mitochondria are present in every nucleated cell in the body. They have a diverse role in cellular metabolism and are the principal source of adenosine triphosphate (ATP). ATP is produced through oxidative phosphorylation (OXPHOS) by the mitochondrial respiratory chain. Impairment of OXPHOS leads to cellular dysfunction and, eventually, cell death. As a result, mitochondrial disorders primarily affect tissues that have a high metabolic demand, such as the neural, muscular, cardiac, ocular and endocrine systems.
Mitochondrial function is dependent on the interplay of mitochondrial DNA and the nuclear genome.2 Mitochondria contain their own DNA (mtDNA), which comprises 16,569 base pairs (bp) and encodes only 37 genes (compared with the 3.3 billion bp in, and thousands of genes encoded by, the nuclear genome). However, most of the approximately 1,500 mitochondrial proteins are coded by nuclear DNA and synthesised on cytoplasmic ribosomes.3 Therefore, abnormalities of mitochondrial proteins can result from defects in both mtDNA and nuclear DNA, and so cause maternally inherited, Mendelian (autosomal dominant, recessive or X-linked), or sporadic diseases (owing to de novo mutations, or recessive inheritance).
How to spot mitochondrial disease
Although mitochondrial disease can present with a heterogeneous array of symptoms, there are certain key neurological clinical features that suggest the possibility of such disease (Fig 1; Table 1). Occasionally, these features form a ‘classic mitochondrial syndrome’ (Table 2). Although some patients fall neatly into a defined syndrome, many patients will be so-called ‘oligosymptomatic’, having one or two components of a syndrome (such as a young individual with an occipital stroke-like lesion, or mild nonfatigable ptosis without ophthalmoplegia), and are more problematic to reach a diagnosis for. These patients often also have several other features that, if present, further support evidence of a mitochondrial disease. A clinician should look and test for these additional feature if suspecting the diagnosis (Table 3). Non-neurological involvement in a patient with unexplained multisystem neurological disease is a ‘red flag’ for mitochondrial disease. Finally, and perhaps most challenging, are patients presenting with ‘non-specific presentations’ (such as epilepsy, diabetes or sensorineural deafness), who often have a multisystem disease that does not fit neatly into any particular category. In these circumstances, the diagnosis of a mitochondrial disorder is often only made in a tertiary referral centre with direct access to specialist laboratory investigations.

Mechanisms of alerting the clinician to the presence of a mitochondrial disorder.
Neurological features that should specifically prompt the clinician to consider a potential mitochondrial disorder.
Classic mitochondrial phenotypes.
Non-neurological symptoms that clinicians should assess for and, if present, add to the suspicion of a mitochondrial disease.
Specific clinical features in ‘suspected mitochondrial disease’
Key neurological features of mitochondrial disease are outlined in Table 1. Chronic progressive external ophthalmoplegia (CPEO) is the most common specific manifestation of mitochondrial disease, and is considered to be present in approximately 20% of patients presenting in adult life.4,5 It typically causes symmetrical limitation of all extraocular muscles and usually occurs in conjunction with ptosis.6 Other patients present with refractory seizure disorders, particularly associated with underlying stroke-like lesions (especially occipital)7 or fluctuating or progressive encephalopathy. Ataxia, peripheral neuropathy and proximal myopathy are common, and can lead to fatigue and myalgia in middle age as the presenting feature.8 Myoclonic jerks, optic atrophy and pigmentary retinopathy also suggest a mitochondrial aetiology. Neurological features, such as extrapyramidal symptoms (Parkinsonism and dystonia) and spasticity, are recognised but uncommon features of mitochondrial disease.
Key extra-neurological features include sensorineural hearing loss developing in young adulthood, short stature, cardiomyopathy, diabetes and other endocrine disorders, often associated with gastrointestinal dysmotility (Table 3).
Classic ‘mitochondrial syndromes’
Several classic clinical syndromes of mitochondrial disease are detailed in Table 2, and have been reviewed elsewhere.9 Some mitochondrial syndromes, such as mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), are readily recognised when ‘full blown’, and suggest specific genetic causes; however, milder forms can be difficult to recognise, particularly in the early stages. Patients usually present after normal childhood development, before sensorineural deafness, diabetes and stroke-like episodes begin during their late teens.10 Genetic blood tests can be misleadingly negative (explained below). Leber's hereditary optic neuropathy (LHON) has a highly stereotypic presentation, with progressive painless visual loss that affects men more than women. Once suspected clinically, LHON is easy to diagnose with a simple blood test, because three mutations (m.3460G>A, m.11778G>A or m.14484T>C) are responsible for 90% of cases.11 Clear diagnostic criteria also exist for Kearns–Sayre syndrome, myoclonic epilepsy with ragged red fibres (MERRF) and mitochondrial neurogastrointestinal encephalopathy (MNGIE) (Table 2).
Family history and heteroplasmy
Mitochondrial disorders can present with any form of inheritance pattern, and one-third of patients have a sporadic disorder.12 Therefore, the importance of taking a family history is to direct genetic tests and to determine potential disease in other family members. If a diagnosis of mitochondrial DNA disease is suspected (and especially if it might be a primary mtDNA disorder; ie there is no male–male transmission in the pedigree), then it is important to ask about subtle clinical features in additional family members.2 This is because different family members often have different phenotypes, owing in part to ‘mtDNA heteroplasmy’.
Heteroplasmy refers to the presence of more than one type of mtDNA genome within a cell. Given that each cell has between 100 and 10,000 mitochondria, each with 2–10 copies of the mtDNA, a mixture of mutated and ‘wild-type’ DNA can exist within cells. Differences in the ratio of mutant and wild-type mtDNA (the ‘mutation load’) between individual family members, and between organs within the same individual, lead to a high degree of clinical heterogeneity. This also poses diagnostic challenges, because low levels of mutant mtDNA in sampled tissues (eg blood) can lead to false negative tests and must always be interpreted with caution. This is particularly the case for the m.3243A>G mutation (see above, in the context of MELAS).
Defining the extent of the clinical phenotype
Several investigations can help to build the ‘mitochondrial phenotype’. Mandatory investigations are an electrocardiogram (ECG), echocardiogram, fasting blood sugar and formal retinal assessment, because manifestations in these systems are often subclinical. Subsequent investigations might be necessary, depending on the phenotype and symptoms (Table 4). Blood lactate levels can be abnormal, especially during an acute metabolic crisis, although lactate levels are often normal in adults presenting with mitochondrial disease subacutely.
Clinical investigations most commonly undertaken to investigate a potential mitochondrial disorder.
Specific mitochondrial investigations
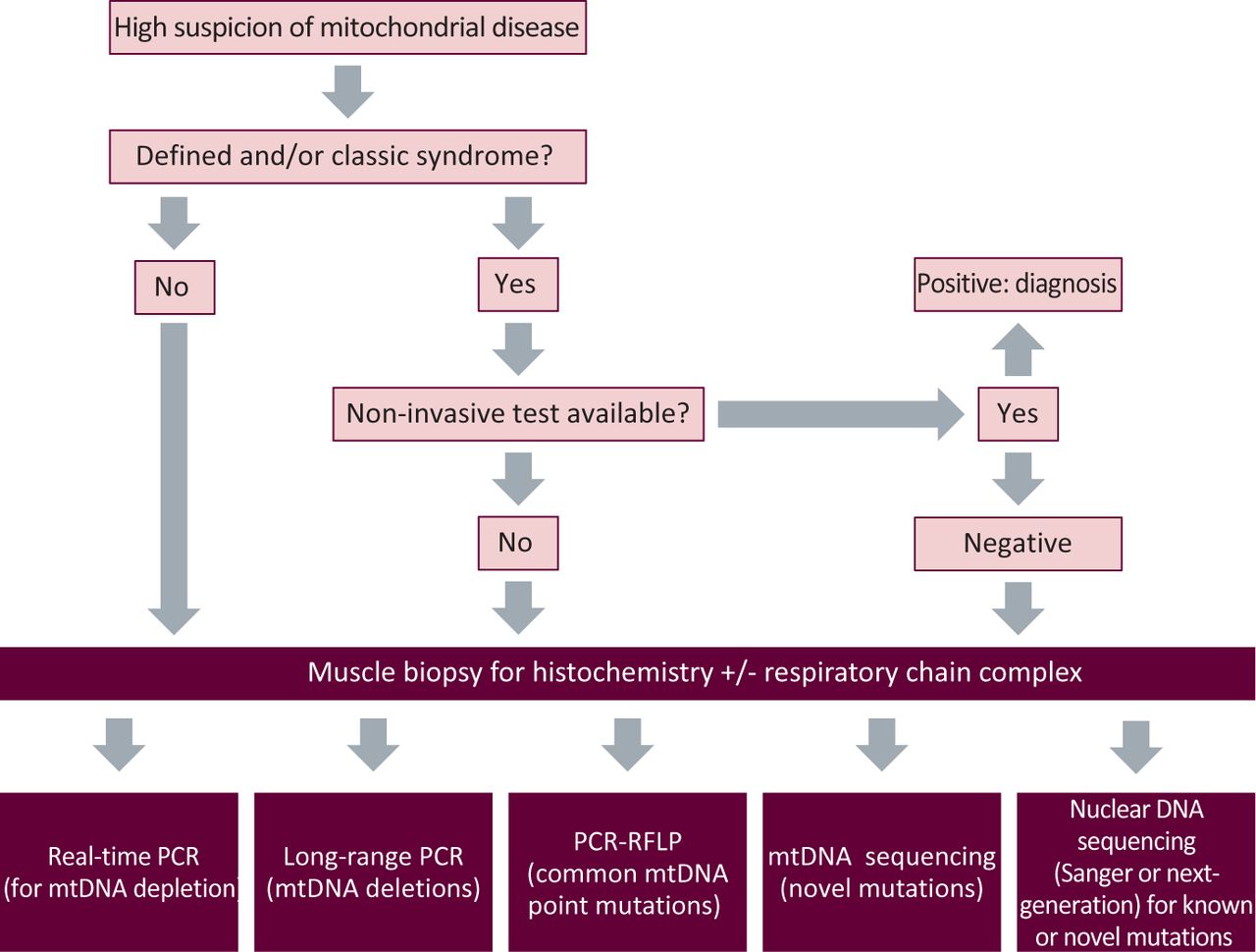
The diagnosis of mitochondrial disorders can range from the simple to awkwardly complex. In the first instance, the decision of whether to embark on this process depends on how strongly the phenotype is felt to represent a mitochondrial disorder. Fig 2 provides an overview of the basic investigative approach.
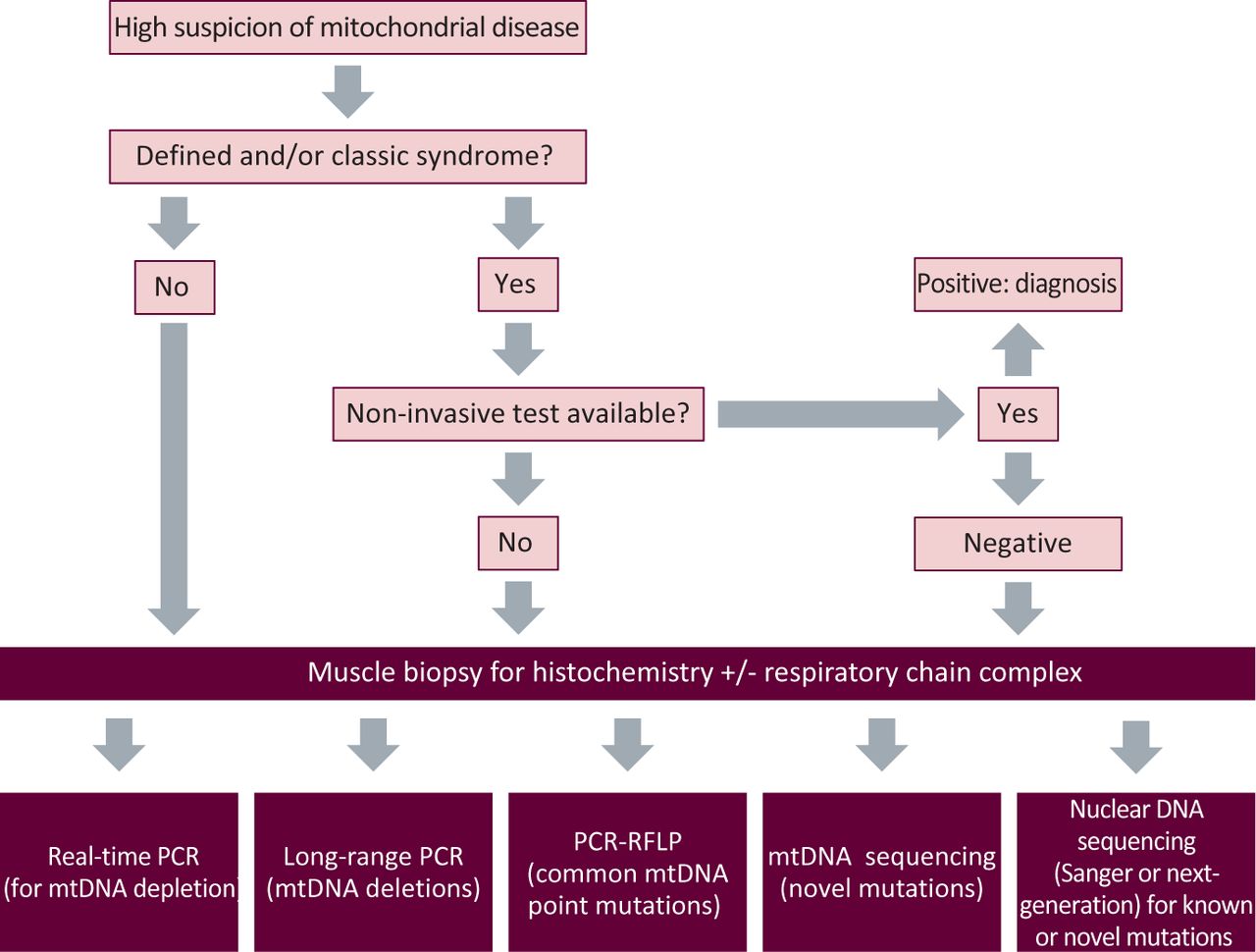
A broad schematic of the investigatory process of suspected mitochondrial disease. mtDNA = mitochondrial DNA; PCR = polymerase chain reaction; RFLP = restriction fragment length polymorphism.
For a specific clinical syndrome, a non-invasive genetic test might be possible on blood, urine or buccal mucosa. However, if these tests return negative, or the clinical features in the first instance necessitate a muscle biopsy (eg CPEO), then this is most commonly undertaken. Other organs are sometimes biopsied (eg liver or myocardium) if they dominate the clinical picture. Histochemical analysis might support a mitochondrial abnormality and guide subsequent biochemical and genetic testing. Thereafter, appropriate testing for mtDNA deletions, depletion, point mutations or nuclear gene screening will lead to a genetic diagnosis in most cases.13,14 However, in a substantial proportion of patients, a genetic diagnosis is not possible using National Health Service (NHS) services. Under these circumstances, it is helpful to engage an active research laboratory with an interest in the patient's phenotype or biochemical abnormality. At present, three laboratories in the UK (in Newcastle, Oxford and London) have been awarded National Specialist Commissioning (NSC) funding to provide the Rare Mitochondrial Disorders Service for Adults and Children, which is a diagnostic and clinical service for patients with mitochondrial disease in England and Scotland.15 With the development of sequencing techniques, such as whole-exome sequencing, this process might change because investigation is likely to become much simpler, less invasive and, ultimately, available across the NHS.
Clinical management
Genetic counselling for mitochondrial disorders is complex. For some mtDNA diseases, such as mtDNA deletions (which often occur as sporadic mutations in patients), the recurrence rate in offspring is low. Other mtDNA disorders are maternally inherited, such as LHON, and have empirical gender-specific recurrence risks that can be given to families. Heteroplasmic mutations are complex and the inheritance pattern can be difficult to predict. Nuclear DNA disorders causing mtDNA disorders are counselled according to their inheritance pattern (dominant, recessive or X-linked), and known clinical penetrance.
Treatment options are also limited for patients, and management remains largely supportive.16 Prevention of secondary complications is best achieved by specialist teams with multidisciplinary resources. Common facets, such as epilepsy, heart failure and gastrointestinal dysmotility, are usually managed according to standard clinical practice, but these features can be challenging to manage in the context of multisystem complications, and long-term follow-up might be required, with ongoing surveillance.
- © 2013 Royal College of Physicians
References








{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.