
Key points
Weak and wasted muscles with retained reflexes is highly suggestive of MND until proven otherwise. Consider the diagnosis when faced with progressive painless weakness in patients over the age of 50.
Consider degenerative spinal disease, especially if the signs (and symptoms) can be attributed to a focal lesion.
Monomelic or bilateral limb onset MND is common, but other more benign and potentially treatable conditions exist and should prompt further assessment.
Beware the diagnosis of MND in patients in whom dysphagia outweighs dysarthria.
Fasciculation without progressive weakness, particularly if symptomatic, is a common and usually benign phenomenon.
Motor neurone disease (MND) is an uncommon (incidence 2–4/100,000 per annum), rapidly progressive neurodegenerative disorder, with onset typically in the sixth and seventh decades (range 20–90+, average age of onset 63 in the UK). It has an invariably fatal outcome, usually from respiratory failure, with 50% of patients dying within 30 months of symptom onset. The diagnosis of MND is clinical, although usually supported by investigations such as electromyography and nerve conduction studies (EMG/NCS), imaging and blood work (see Table 1).1–3
Investigation of suspected MND.
Amyotrophic lateral sclerosis (ALS) is the most common form of MND, and is characterised by the often synchronous degeneration of upper motor neurones (UMN) and lower motor neurones (LMN). Other clinical phenotypes exist, including a pure UMN variant (primary lateral sclerosis, PLS), a pure LMN variant (progressive muscular atrophy, PMA) and an initially isolated bulbar muscle variant (progressive bulbar palsy, PBP). Diagnostic criteria for MND exist, but these are not useful in routine clinical practice. The significant variation in site and speed of onset and progression often leads to diagnostic delay.4 Furthermore, given the gravity of the diagnosis, many physicians are hesitant to make it fearing both a difficult consultation and the possibility of missing an alternative and potentially treatable condition. With increasing specialisation, it is now best practice that the diagnosis of MND be made by a neurologist with knowledge of and access to appropriate support services.
Classical ALS phenotype of MND
The key presenting feature of MND is progressive, painless weakness, and thus the list of potential differential diagnoses is long (see Table 2). Despite this, and the clinical variability discussed above, the misdiagnosis rate for MND is relatively low at 7–8%.5,6
Mimic syndromes considered in the differential diagnosis of MND.
The most common ALS mimic is degenerative spondylotic myeloradiculopathic disease of the cervical and/or lumbo-sacral spine. Patients with degenerative disease often have pain, which may be radicular. Although initial symptoms of spondylotic disease might be progressive, they may plateau, a feature not seen in MND.7 Imaging will help to identify patients who have spondylotic disease, although as a further complication, many ALS patients will also have radiological evidence of degenerative spinal disease, which is likely to be clinically irrelevant.
Lower motor neurone syndromes affecting the limbs
Misdiagnosis is more common when the symptoms affect only the LMN, with a diagnostic error rate for MND approaching 20%.8 Importantly, several MND mimics are potentially amenable to treatment or have a significantly more benign prognosis (see Table 3). The prompt referral of such patients to specialist neurological services for assessment is thus important. Even in expert hands, serial assessment is often required to allow accurate diagnosis, and the importance of clinical follow up cannot be overstated.
Generalised or focal single (monomelic) limb onset lower motor neurone syndromes mimicking MND.
Generalised weakness
Kennedy's syndrome9,10 (also known as X-linked spinobulbar muscular atrophy) can present as predominantly distal LMN weakness in the limbs and should be considered in the differential, particularly as bulbar dysfunction often follows limb involvement by many years10 (Table 3). Pure motor forms of chronic inflammatory demyelinating polyneuropathy (CIDP) occur and are the reason why some specialists advocate a trial of intravenous immunoglobulin therapy in pure LMN conditions.11 Distal and inclusion body myopathies (IBM) should also be considered (see below and Table 3).12,13
Focal onset limb (regional) syndromes
The focal onset of weakness in a single limb (monomelic disease, typically LMN in character) is a common presentation of MND. In this setting, however, the differential diagnosis is broad and includes a number of potentially treatable or more benign conditions (such as multifocal motor neuropathy with conduction block14) (Table 3). The key to the correct diagnosis of MND is the recognition of progressive weakness and the evolution of physical signs of mixed motor system degeneration (i.e. UMN and LMN signs in one or more limb).
Bilateral lower limb LMN paresis
Although an increasingly recognised and relatively benign MND phenotype (historically referred to as the polyneuritic variant of MND or ascending paralysis), such a presentation can lead to significant diagnostic delay. Lumbo-sacral degenerative spondolytic disease, pure motor neuropathy/neuronopathy11 and radiation (myelo) radiculopathies15 are included in the list of differential diagnoses, as are distal myopathies and IBM. Detailed investigation and follow up, with serial EMG/NCS examinations and sometimes muscle biopsy should ultimately lead to accurate diagnosis.
Bulbar and corticobulbar syndromes
Of patients with MND, 20–25% will present with bulbar dysfunction. Typically, such bulbar onset MND patients will develop symptoms and signs of limb involvement within 12 months of bulbar symptoms.1–3 Almost without exception, the dysarthria associated with MND precedes and outweighs any dysphagia.
Structural disease
Where dysphagia exceeds dysarthria, clinicians should be prompted into a careful search for an alternative diagnosis. In my experience (of more than 800 incident cases of MND), such a presentation (dysphagia greater than dysarthria) of MND has occurred only twice. The patient represented in Fig 1a was referred by gastroenterology with dysphagia of unknown cause (but no dysarthria). The unusual history prompted a request for imaging.
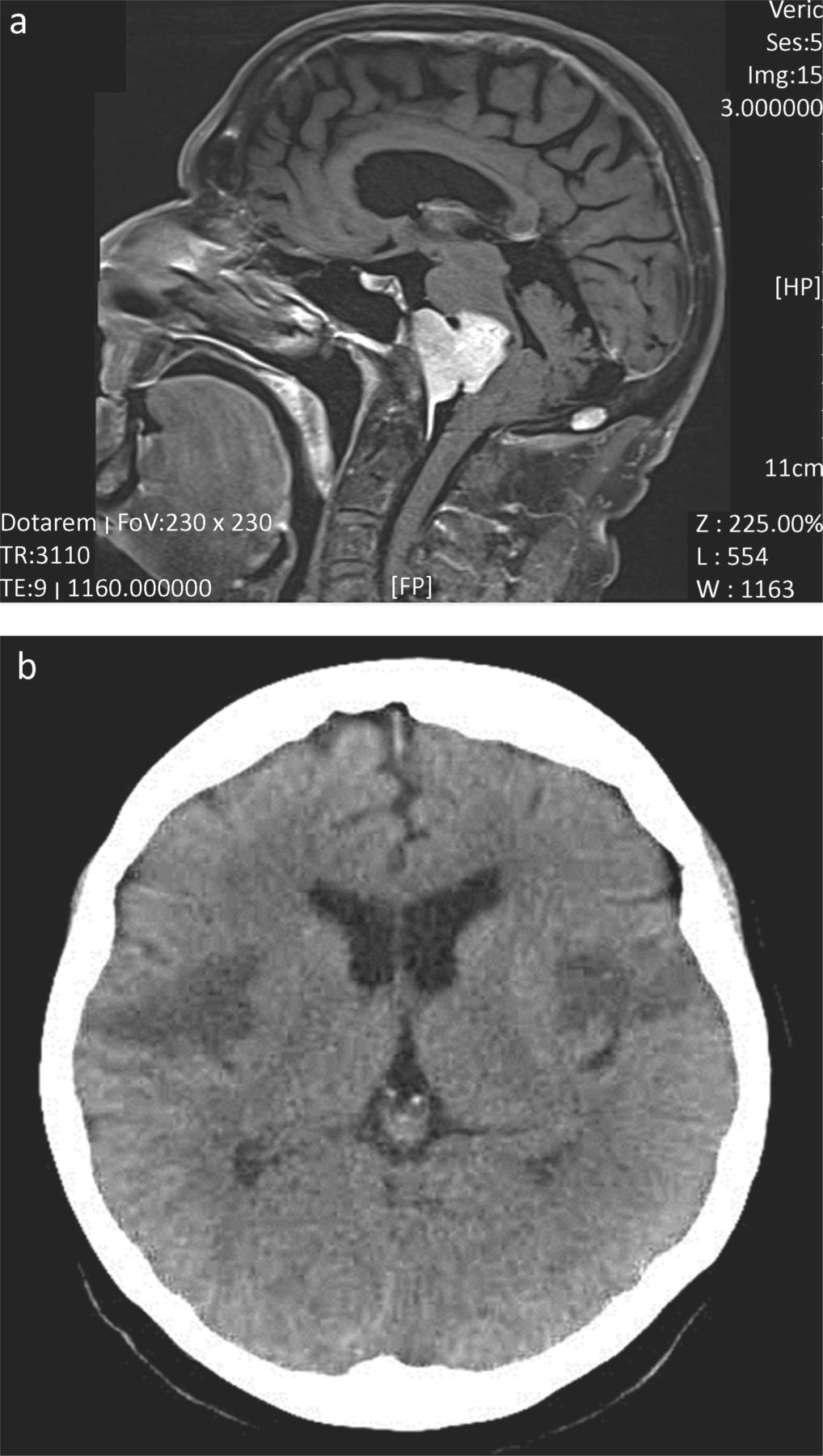
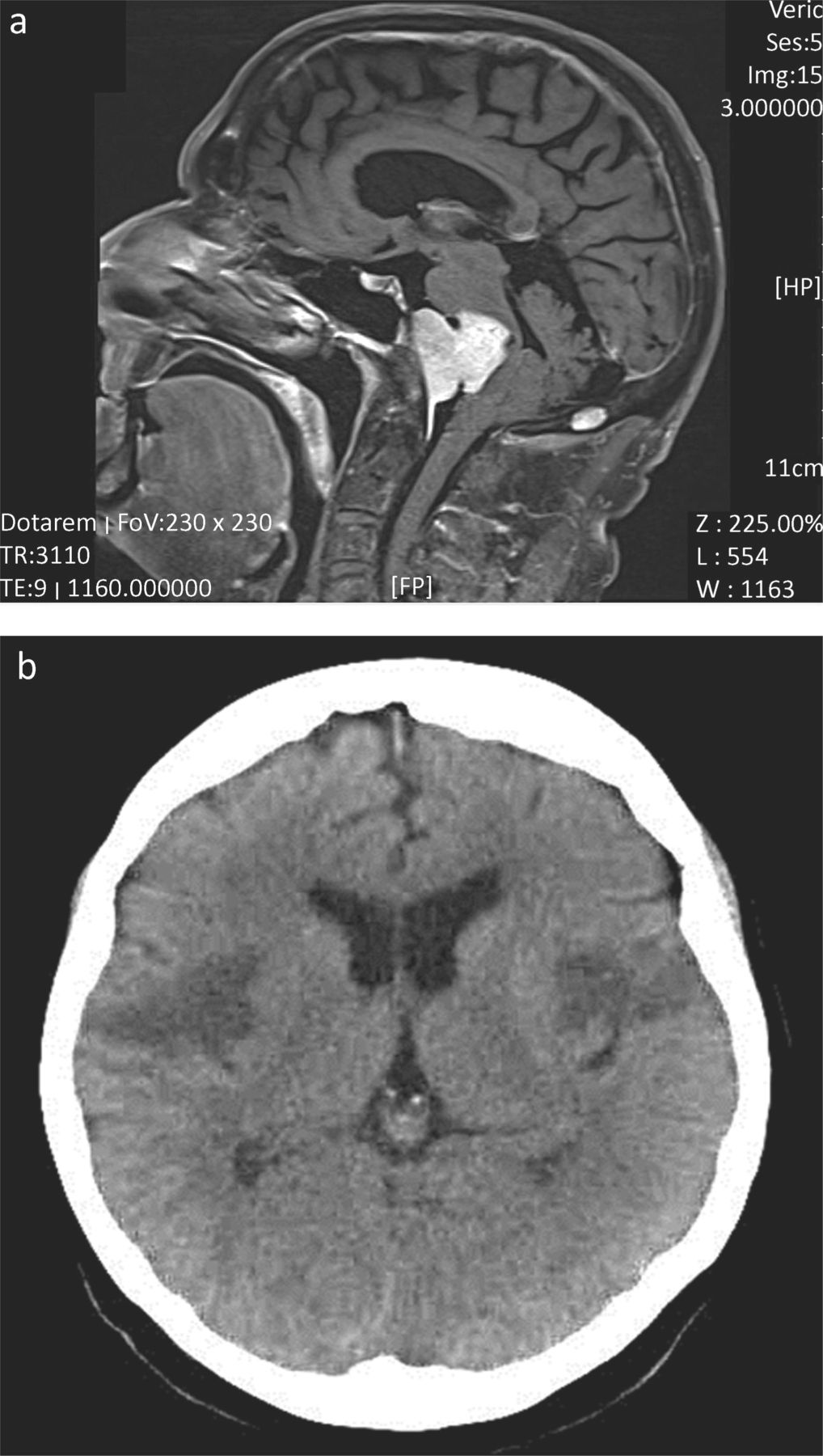
Imaging of patients with dysphagia greater than dysarthria. (a) Midline T2 weighted sagittal MR head. This patient presented with 12 months of increasing dysphagia without dysarthria. There were no abnormalities on examination. Extensive assessment by gastroenterology, ENT, upper gastro-intestinal surgery and investigation (endoscopic examination, barium swallow and blood work) had failed to yield a diagnosis. MR imaging was requested to exclude a structural lesion as a cause for dysphagia. The imaging demonstrated a large midline clival meningioma, which was successfully removed by surgery. The patient has a residual left facial palsy, but near normal swallow. (b) Representative non-enhanced CT brain head from a patient who presented with the abrupt onset of mild dysphasia and right-sided weakness, followed over the next 48 hours by complete aphagia. Although initial CT imaging demonstrated infarction of the left operculum, repeat CT imaging 24 hours later (shown here) demonstrated bilateral infarcts representing the opercular or Foix–Chavany–Marie-syndrome (dysphagia, dysarthria, drooling and facial weakness, often with limb sparing), which is seen as a result of stroke, infection and occasional neurodegenerative disease. Supportive treatment and temporary naso-gastric tube feeding resulted in a good functional recovery.
Vascular disease
Diffuse vascular disease is often considered in the differential diagnosis of MND, although such patients will typically have symptoms and signs beyond that of a pure motor system disorder. MND can be initially misdiagnosed as a stroke, but the progression of symptoms should prompt reconsideration, and emphasises the importance of follow up. Focal vascular disease in the brainstem is very unlikely to result in isolated dysarthria and dysphagia, and will usually be associated with long tract (sensory and/or motor) signs. Occasional rare vascular syndromes can cause diagnostic difficulty. Fig 1b illustrates a patient who presented with sub-acute (over two days) dysphagia and dysphasia. The diagnosis, confirmed on imaging, was of the opercular or Foix–Chavany–Marie syndrome.
Other neurodegenerative conditions can cause dysphagia and, although rare, can lead to misdiagnosis (e.g. progressive supranuclear palsy and Lewy body disease16).
Neuromuscular transmission (NMT) disorders
The most common NMT disorder in the context of isolated or near isolated bulbar dysfunction (there can be mild limb girdle weakness in NMT disease and MND) is myasthenia gravis (MG). Characterised by fatigable weakness of ocular, oculo-bulbar and/or limb girdle muscles, MG is occasionally misdiagnosed as MND and vice versa. Muscle fatigue, although considered a characteristic feature of MG, occurs in patients with other neuromuscular disorders including MND. Brain imaging, EMG/NCS and anti-acetylcholine receptor autoantibodies (which are negative in 10–15% of patients with generalised MG) should allow accurate diagnosis. EMG/NCS might not be diagnostic of MND in bulbar onset disease,3 but should allow the exclusion of other conditions affecting NMT, such as MG. Pyridostigmine, a cholinesterase inhibitor used for the treatment of MG, can provide transient symptom relief in MND.3,17
Fasciculation syndromes
Widespread, asymptomatic fasciculation (of which the patient is unaware other than by observation) is highly suggestive of MND. Focal episodic symptomatic fasciculation, typically affecting just one or a select group of muscles without progressive weakness, is common, however, and unlikely to represent disease. When fasciculation is persistent, it can represent benign muscle cramp or fasciculation syndrome. Patients can be reassured, although EMG/NCS might be required.18,19 Certainly muscle fasciculation without weakness should be considered a benign phenomenon, although observation over time (sometimes 6 months or more), might be required to confirm that conclusion.
Summary
The misdiagnosis of MND (particularly of the ALS phenotype), is uncommon. Atypical presentations, particularly of focal onset and with pure LMN or UMN signs, present a more difficult diagnostic challenge, although perhaps reassuringly, treatable mimics are rare. A working knowledge of potential alternative conditions and MND diagnostic pitfalls should help to reduce the misdiagnosis rate, particularly if the key points are considered.
- © 2013 Royal College of Physicians








{kind=link}