
Key points
Myeloproliferative neoplasms (MPN) are primary, clonal expansions of myeloid lineage cells (erythrocytes, platelets and granulocytes)
MPN are often associated with mutations or translocations in the genes that regulate cell proliferation
A mutation in the JAK2 gene is found in more than 97% of polycythaemia vera (PV) and approximately 60% of essential thrombocythaemia (ET) and primary myelofibrosis (PMF) patients, and has revolutionised diagnosis of these conditions.
The major risk associated with PV and ET is arterial and venous thrombosis. Aspirin and control of the red cell and/or platelet count with venesection or cytoreductive drugs greatly reduce the risk of thrombotic events.
Inhibitors of JAK2 are showing early promise in the treatment of PMF, the most aggressive of the MPN.
The myeloproliferative neoplasms (MPN) are a group of clonal haematological diseases, whose characteristic feature is proliferation of one or more of the myeloid lineages (Fig 1). The four disorders that are most commonly included under MPN are listed in Table 1. Evidence is emerging that shows MPN is the result of various acquired mutations, which permanently activate genes (often coding for tyrosine kinases) that regulate cell proliferation. These mutations are better defined for some conditions than others. The more commonly encountered MPN and their associated lesions are shown in Table 1.
Common mutations involved in MPN disorders.

Haematopoiesis and the lineages that are predominantly affected in MPN. Myeloid haematopoiesis derives from pluripotent myeloid stem cells in the marrow, which give rise to the erythroid, megakaryocyte (platelet), granulocyte (neutrophil, eosinophil and basophil) and monocyte lineages. The point in this hierarchy at which MPN-associated mutations occur could partially determine which cells are predominantly over-produced, giving rise to the different clinical phenotypes of the MPN. BFU = burst-forming unit; CFU = colony-forming unit; E = erythroid; Meg = megakaryocytic; GM = granulocyte-monocyte; G = granulocytic; M = monocytic; Eo = eosinophilic; Ba = basophilic.
Of these, chronic myeloid leukaemia (CML) is the best understood at a molecular level. The translocation known as Philadelphia chromosome t(9;22) results in the ABL gene from chromosome 9 being transferred to chromosome 22. The product of this translocation (BCR-ABL) is an oncoprotein that has increased tyrosine kinase activity, which results in deregulated proliferation of leukaemic myeloid cells. Targeted treatment (with tyrosine kinase inhibitors such as imatinib) blocks BCR-ABL and produces very durable remissions (now of more than 10 years) in the majority of patients with CML — a huge improvement in outcome for this condition.
The MPN most commonly seen by non-haematologists — essential thrombocythaemia (ET), polycythaemia vera (PV) and primary myelofibrosis (PMF) — are those associated with the Janus-associated kinase 2 (JAK2) V617F mutation. JAK2 is a signalling protein whose normal role is to activate cell proliferation following the binding of erythropoietin to its receptor (Fig 2). The activating V617F mutation leads to a constant ‘on switch’ that drives proliferation of myeloid precursors. How a single mutation results in three clinical phenotypes (and how some patients have the disease without the mutation) is likely to depend on the roles of other, some as-yet-unidentified, mutations and is currently under investigation.1
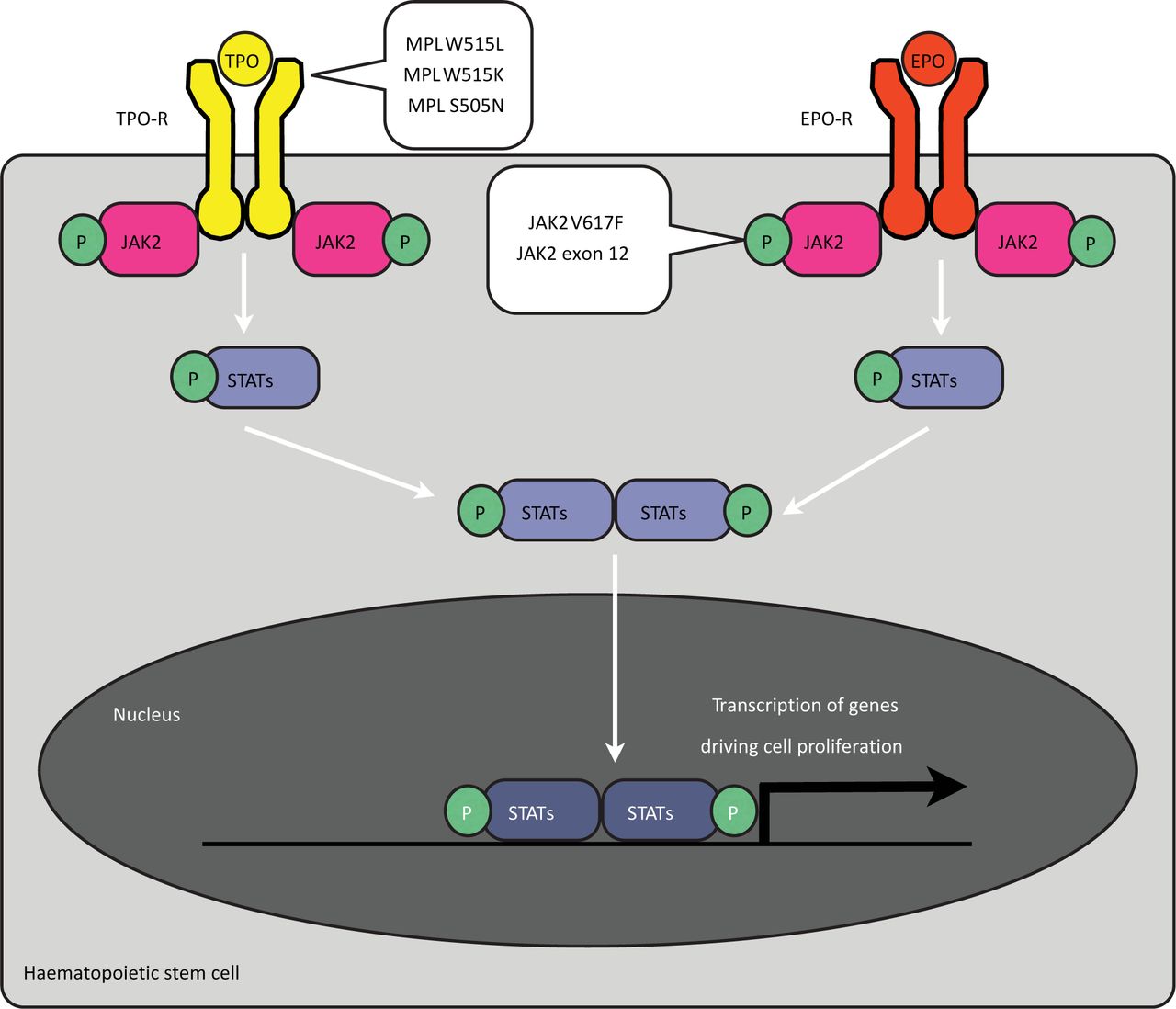
Regulation of haematopoiesis by the erythropoietin (EPO) and thrombopoietin (TPO) receptors (EPO-R and TPO-R). The erythrocyte and platelet growth factors erythropoietin and thrombopoietin stimulate cell growth by binding cell surface receptors on haematopoietic progenitor cells. Receptor ligation leads to phosphorylation of the JAK2 tyrosine kinase and recruitment and activation of STATs. Activated, dimerised STATs pass into the nucleus, where they regulate the expression of genes that are required for cell-cycle activation and cell proliferation. Mutations in the JAK2 gene that result in constitutive activity are associated with PV, ET and PMF. Mutations in the MPL gene, coding for the TPO-R, are found in ET. CML = chronic myeloid leukaemia; EPO = erythropoietin; ET = essential thrombocythaemia; JAK2 = Janus-associated kinase 2; MPL = myeloproliferative leukemia; P = phosphorous; PMF = primary myelofibrosis; PV = polycythaemia vera; STAT = signal transducers and activators of transcription; TPO = thrombopoietin.
Polycythaemia vera
Polycythaemia refers simply to a raised haematocrit (HCT). It is important to note that the diagnosis is based on the haematocrit and not the haemoglobin concentration. In PV, the cause of the raised HCT is a primary marrow disorder resulting in red cell overproduction.
Polycythemia might also be secondary or apparent. In secondary polycythemia, excess red blood cells are produced in response to an increase in erythropoietin concentrations in the blood. In turn, an increase in erythropoietin can be seen in patients with chronic hypoxia (from congenital cyanotic heart disease or severe pulmonary disease) or, occasionally, because of excess erythropoietin production resulting from a tumour, most commonly renal cell cancer.
In apparent or relative polycythemia the red cell mass is normal but the plasma volume is reduced from causes such as diuretic use, obesity, smoking or alcohol excess (Table 2).
Differential diagnosis of polycythaemia and thrombocytosis.
These alternative causes of polycythemia are not discussed further here. Distinguishing between PV and the other possible causes of a raised haematocrit is important because the outcome in PV is improved by treatment that reduces the haematocrit, whereas there is little evidence for venesection in secondary polycythaemia. As almost 100% of patients with PV are positive for a JAK2 mutation, diagnosis has become much simpler in recent years.
The incidence of PV is 0.7–2.6 per 100,000 per year, with a slight male predominance (M:F, 1.2:1) and a median age of 55–60 years.
Investigation
For a diagnosis of polycythemia, HCT should be >0.52 in men and >0.48 in women. A careful history and examination are often sufficient to distinguish patients who are likely to have PV from those with a probable secondary cause. Initial investigations should include a full blood count (FBC) and film, a JAK2 V617F screen, ferritin level and basic biochemistry.
Clinical features and complications
Patients with PV might complain of pruritis (aquagenic, following a hot bath or shower), neurological symptoms (headaches or drowsiness) or gout. Examination commonly reveals a plethoric appearance and splenomegaly. The major morbidity in PV is the result of thrombosis (including mesenteric and hepato-portal thromboses). Splanchnic vein thrombosis is frequently associated with PV and can occur before a raised HCT is present. An unprovoked splanchnic vein thrombosis should prompt JAK2 testing.
Treatment
First-line treatment for PV remains venesection, with a target HCT of <0.45. Aspirin (75 mg) should be given providing there is no contraindication. Cytoreduction should be considered when the patient is aged >60 years or is unable to tolerate venesection, or where there is thrombocytosis, systemic symptoms or symptomatic splenomegaly. First-line treatment is generally with interferon for those of <40 years old and with hydroxycarbamide for those >40 years old.
Prognosis
In the past, many patients died within a few years of diagnosis as a result of thrombotic complications. Median survival is now greater than 10 years. The greatest risk is still from thrombosis. Of patients treated with venesection alone, 2–3% will progress to myelodysplastic syndrome (MDS) or acute myeloid leukaemia (AML). The risk of progression to myelofibrosis is 10–15% at 10 years.
Essential thrombocythaemia
Essential thrombocythaemia is a disorder of the megakaryocyte lineage. Given the prevalence of reactive thrombocytosis, diagnosis is often difficult. In the absence of the JAK2 V617F or other clonal mutation, it is essentially one of exclusion.
To meet the World Health Organisation (WHO) 2008 guidelines for diagnosis,2 the patient must meet all four of the following criteria:
sustained (during the time of investigation) platelet count ≥450 × 109/l
bone marrow biopsy showing proliferation of megakaryocytes with increased numbers of enlarged mature megakaryocytes
failure to meet the WHO criteria for other myeloproliferative neoplasms
carry either the JAK2 V617F mutation (present in about 50% of cases) or another clonal marker, or have no evidence of reactive thrombocytosis (see Table 2).
Incidence is estimated at 0.6–2.5 per 100,000 per year. There is no sex predominance and the majority of cases present in patients aged 50–70 years.
Clinical features
Thrombosis is the major cause of morbidity and mortality for those with ET (the precise rate is not known, but has been estimated at approximately 25% at two years in untreated, high-risk patients) and can be either arterial or venous. Age of more than 60 years and a previous history of thrombosis both further increase the thrombotic risk. Paradoxically, haemorrhage can also occur, especially in those whose platelet count is >1,000 × 109/l. Increased platelet numbers bind von Willebrand's factor, resulting in increased clearance and a form of acquired von Willebrand's disease.
Treatment
Patients with ET can be stratified into high-, intermediate- and low-risk groups, and their disease management is dependent on risk.3
High risk: aged >60 years or platelets >1,500 × 109/l, an ET-related thrombotic or haemorrhagic event.
Intermediate risk: aged 40–60 years, no other high-risk factors.
Low risk: aged <40 years, no risk factors.
Other cardiovascular risk factors should be aggressively managed for all ET patients. First-line treatment for high-risk patients is hydroxycarbamide and aspirin. PT-1,4 the largest randomised study of ET, showed superiority for treatment with hydroxycarbamide and aspirin over anagrelide and aspirin in terms of a reduced risk of arterial thrombosis, major haemorrhage and transformation to myelofibrosis, but not venous thromboembolism. The benefit of treatment for the intermediate risk group is less clear. The majority of patients are treated with hydroxycarbamide and aspirin. Low-risk patients can generally be managed with aspirin alone.
Prognosis
Most patients will have long, symptom-free episodes, with the major risk being thrombosis or haemorrhage. There is a risk of transformation, to either myelofibrosis or acute myeloid leukaemia (AML); risks at 10 years are <10% and approximately 3%, respectively. Because of the usual age of onset, however, life expectancy is often near normal.
Primary myelofibrosis
In PMF, as in ET, the clonally proliferating cells are mostly abnormal megakaryocytes. The release of cytokine derived from the clone causes reactive deposition of fibrous connective tissue, leading to the characteristic features of the disease. Replacement of the normal bone marrow space with fibrous tissue prevents normal haematopoiesis and leads to anaemia and, eventually, pancytopenia (although peripheral blood counts, especially platelet counts, can be high early in the disease). Abnormal, ineffective extramedullary haematopoiesis is probably a partial attempt to correct for the loss of marrow function and often leads to massive splenomegaly. Systemic features, secondary to cytokine release, are common and include fevers, weight loss, anorexia and sweats. The replacement of the marrow by fibrosis results in the displacement of immature cell forms, such as nucleated red cells and myelocytes, into the peripheral blood (a leucoerythroblastic picture). The passage of normal erythrocytes through the narrowed fibrotic marrow spaces distorts them, generating the classic ‘tear-drop’-shaped red cells.
Incidence is equal in both sexes at 0.5–1.5 per 100,000 per year and the most common age of onset is 50–60 years. In addition to the primary disease, myelofibrosis can also occur secondary to ET or PV. Myelofibrosis can also be a feature of other malignancies, such as Hodgkin lymphoma, when there is marrow involvement.
Investigation
Full blood count and film are as described above. The JAK2 V617F mutation is present in approximately 60% of patients. Liquid bone marrow often cannot be aspirated because of the fibrosis. Trephine biopsy typically shows disorganisation of the normal marrow architecture, with increased numbers of morphologically abnormal megakaryoctyes. Reticulin staining identifies the fibrous tissue and is increased (especially later in the disease process).
Treatment
The main treatment for PMF has been supportive therapy (blood product support, rapid treatment of infection). Hydroxycarbamide might be needed in the early proliferative phase to control the white blood count or to reduce symptomatic splenomegaly.
In younger patients who are fit for the procedure, allogeneic stem cell transplantation offers a potentially curative treatment by replacing the abnormal clonal marrow with healthy donor haematopoietic stem cells. Regression of bone marrow fibrosis is observed (over time) following successful allogeneic transplantation. For patients who are unfit for transplant or without a potential donor, the new JAK2 inhibitors (such as ruxolitinib) are showing promise.
Prognosis
PMF carries the poorest prognosis of the MPN. Median survival is estimated at 5–7 years, but is dependent on the stage at which the disease is diagnosed, with some patients surviving 10–15 years and others less than 12 months. Death results from bone marrow failure (infection or bleeding), thromboembolism, cardiac failure or transformation to acute leukaemia.
Summary
The myeloproliferative neoplasms that are associated with the JAK2 mutation are a heterogeneous group of disorders. The additional mutations that result in the clinical phenotype are still the subject of research. As more than one mutation is involved, and as JAK2 has a necessary physiological role (unlike BCR-ABL), the development of targeted therapy remains a challenge. Although new drugs are being developed, treatment at present is predominantly with agents that have been in use for many years. An understanding of the need to control the thrombotic risk has, however, led to improved survival rates such that ET and PV can be seen as chronic diseases.
Acknowledgments
We would like to thank Dr Andrew Duncombe and Dr Nancy Colchester for helpful comments on the manuscript, and Professor Nick Cross for assistance with the figures.
- © 2013 Royal College of Physicians








{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.