
Key points
Genetic advances have enabled redefinition of diseases according to their true causes and this molecular-based taxonomy has transformed our understanding of medicine and improved diagnosis and treatments for patients1
Genetic testing is helpful in clinical practice in several ways, including:2 confirmation of the clinical diagnosis; identification of asymptomatic family members who harbour the mutation and require screening for the development of early disease manifestions, thereby facilitating earlier treatment; and identification of family members who do not harbour the familial germline mutation and can therefore be reassured and alleviated of the burden of anxiety of developing the disease
Such genetic testing can be performed either postnatally or prenatally depending on the disorder and the treatments available
Elucidation of the genetic cause has enabled the identification of targets for therapy, which is illustrated by the use of tyrosine kinase inhibitors to target activated tyrosine kinase receptors that result from RET mutations that cause medullary thyroid cancer
All patients undergoing genetic testing should have access to genetic counselling
Introduction
Significant advances in molecular genetics have enabled the genetic abnormalities associated with endocrine disorders to be identified and thereby facilitated the redefinition of diseases according to their true causes.1 This molecular-based taxonomy has transformed our understanding of medicine and improved diagnosis and treatment for patients.1 This article reviews some of these advances that have had an impact on the diagnosis and management of endocrine disorders, using examples from the multiple endocrine neoplasia syndromes and congenital adrenal hyperplasia. Table 1 gives a list of some endocrine conditions that have a genetic basis.
Some endocrine disorders with associated genetic abnormalities.*
Role of mutation testing in clinical practice
Mutational analysis in hereditary endocrine disorders and complex syndromes is helpful in clinical practice in several ways, including confirmation of the clinical diagnosis; identification of asymptomatic family members who harbour the mutation and require screening for the development of early disease manifestations, thereby facilitating earlier treatment; and identification of family members who do not harbour the familial germline mutation and can therefore be reassured and alleviated of the burden of anxiety about developing the disease.2 This latter aspect cannot be overemphasised because it helps to reduce the costs to both individuals and health services in terms of eliminating the need for unnecessary repeated biochemical and radiological investigations.2 Mutational analysis thus can be useful in clinical practice, which is illustrated with reference to postnatal testing in the context of multiple endocrine neoplasia syndromes and prenatal testing in the context of congenital adrenal hyperplasia.
Multiple endocrine neoplasia
Multiple endocrine neoplasia (MEN) is characterised by the occurrence of tumours involving two or more endocrine glands within a single patient.2 The two major forms of MEN are type 1 (MEN1, Wermer’s syndrome) and type 2 (MEN2, Sipple’s syndrome) (Table 2). MEN1 is characterised by the predominant occurrence of parathyroid, pancreatic islet and anterior pituitary tumours. MEN2 is characterised by the occurrence of medullary thyroid carcinoma (MTC) in association with phaeochromocytomas, with three clinical variants referred to as MEN2A, MEN2B and MTC only (Table 2). MEN1 and MEN2 are autosomal dominant disorders due to mutations in the tumour suppressor gene MEN1, which encodes the 610-amino acid protein menin, and the rearranged during transfection (RET) proto-oncogene, which encodes a 1,114 amino acid tyrosine kinase receptor (TKR), respectively. The finding of MEN1 or MEN2 in a patient has important implications for family members, because first-degree relatives have a 50% risk of developing the disease, but individuals with and without the disease can be identified by mutational analysis.2
Multiple endocrine neoplasia syndromes and their associated tumours. Adapted with permission from Thakker et al.2
Multiple endocrine neoplasia type 1
Diagnosis. Mutational analysis of MEN1 should be undertaken in:
an index case with two or more MEN1-associated endocrine tumours (that is, parathyroid, pancreatic or pituitary tumours)
asymptomatic first-degree relatives of a known carrier of MEN1 mutations
first-degree relatives of a carrier of MEN1 mutations expressing familial MEN1 (that is, having symptoms, signs and biochemical or radiological evidence for one or more MEN1-associated tumours)
patients with suspicious or atypical MEN1, which includes individuals with parathyroid adenomas occurring before the age of 30 years or multigland parathyroid disease, gastrinoma or multiple pancreatic neuroendocrine tumours (NET) at any age, and individuals who have two or more MEN1-associated tumours that are not part of the classic triad of parathyroid, pancreatic islet and anterior pituitary tumours.2
Management and treatment. Mutational analysis should be undertaken within the first decade in children who have a parent or first degree relative with MEN1, because children with MEN1 tumours have been reported by the age of 10 years and appropriate intervention in the form of biochemical testing or treatment, or both, should be considered.2 A genetic test confirming that an individual who may be an asymptomatic relative of a patient with MEN1 is a mutant gene carrier is likely to lead to earlier and more frequent biochemical and radiological screening rather than immediate medical or surgical treatment. In contrast, those relatives who do not harbour the MEN1 mutation have a markedly reduced risk of developing MEN1-associated endocrine tumours, equivalent to that of the general population, thereby freeing these relatives without the MEN1 mutation from the requirement for further repeated clinical investigations. Identification of MEN1 mutations thus may be of help in the clinical management of patients and their families with this disorder. Mutational analysis of MEN1 in a symptomatic family member (that is, an individual already showing a clinical manifestation of MEN1) from a family with a known MEN1 mutation has been challenged as being unnecessary to establish the diagnosis of MEN1. However, studies have reported that phenocopies that may confound the diagnosis occur in 5–10% of MEN1 kindreds.2 MEN1 family members with one MEN1-associated tumour therefore should be offered mutational analysis of MEN1. Mutant gene carriers should be screened at appropriate 1–3-year intervals for primary hyperparathyroidism (using albumin-corrected calcium and parathyroid hormone (PTH)), pancreatic NET (using fasting gastrointestinal hormones and magnetic resonance imaging (MRI)), pituitary tumours (using prolactin, insulin-like growth factor-1 and MRI of the pituitary gland), adrenal tumours (using MRI), and thymic and bronchial carcinoid tumours (using computed tomography (CT) or MRI).The treatments for each MEN1-associated tumour are broadly similar to those for non-MEN1-associated tumours, although there are important differences which are reviewed in the clinical practice guidelines for MEN1.2
Multiple endocrine neoplasia type 2
Diagnosis. Mutational analysis of RET should be undertaken in
an index case with MTC, MEN2 or primary C cell hyperplasia
all individuals with a family history of MEN2 or familial MTC (FMTC)
first-degree relatives of a known carrier of a RET mutation
patients with MEN2-associated features such as intestinal ganglioneuromatosis, lichen planus amyloidosis (which may be associated with pruritus in the central upper back) and Hirschsprung’s disease.3
Mutational analysis of RET should also be undertaken in at-risk children at the earliest possible age and definitely before 5 years of age. Prenatal testing, using chorionic villus sampling or amniocentesis in the first and second trimesters, respectively, is available for mutational analysis of RET, if required.
Management and treatment. Ten-year survival in patients with metastatic MTC is about 20%,4 and prophylactic thyroidectomy to prevent MTC and its metastases is recommended in patients with a RET mutation, especially as the onset of MTC is earlier in the hereditary forms, with foci of MTC arising in infancy.3 Children with MEN2B-associated mutations – RET mutation Met918Thr (>95% of MEN2B mutations), rarer Ala883Phe RET mutations and double RET mutations (Val804Met + Glu805Lys, Val804Met + Tyr806Cys, and Val804Met + Ser904Cys) – should undergo prophylactic thyroidectomy as soon as possible and within the first year of life; prophylactic thyroidectomy in children with MEN2A- or FMTC-associated RET mutations may be delayed until they are aged 5 years.3 Such prophylactic thyroidectomy has dramatically improved outcomes in patients with MEN2, such that about 90% of young patients with a RET mutation who have had a prophylactic thyroidectomy have no evidence of persistent or recurrent MTC at 7 years after surgery.5,6 Screening for MEN2-associated tumours in carriers of gene mutations should be undertaken annually in the form of neck ultrasound and basal and stimulated plasma calcitonin measurements for MTC; albumin-corrected calcium or ionised calcium with PTH for primary hyperparathyroidism; and 24-hour urinary catecholamine measurements for phaeochromocytoma.
Tyrosine kinase inhibitors in the treatment of advanced MTC. The finding that RET encodes a tyrosine kinase receptor (TKR) and that MEN2-associated mutations result in activation of the TKR indicated that it might be a target for the treatment of advanced or metastatic MTC, which responds poorly to radiotherapy and chemotherapy.3,7 Tyrosine kinase inhibitors (TKIs) (Table 3)8–14 thus have been used to treat familial MTC and non-familial MTC, 70% of cases of which have a somatic RET mutation.15 The TKI vandetanib has been used to treat rapidly progressive MTC, with >80% of patients reported to have 6-month progression-free survival, compared to 60% with placebo.16 Another TKI, cabozantinib, which is licensed for use in advanced, rapidly progressive MTC, has been reported to result in a median progression-free survival of >11 months compared to 4 months with placebo.17 The evidence for a role of TKIs for treatment of MTC thus seems promising.
Treatment outcomes for advanced medullary thyroid cancer (MTC) using oral tyrosine kinase inhibitors (TKIs).
Congenital adrenal hyperplasia
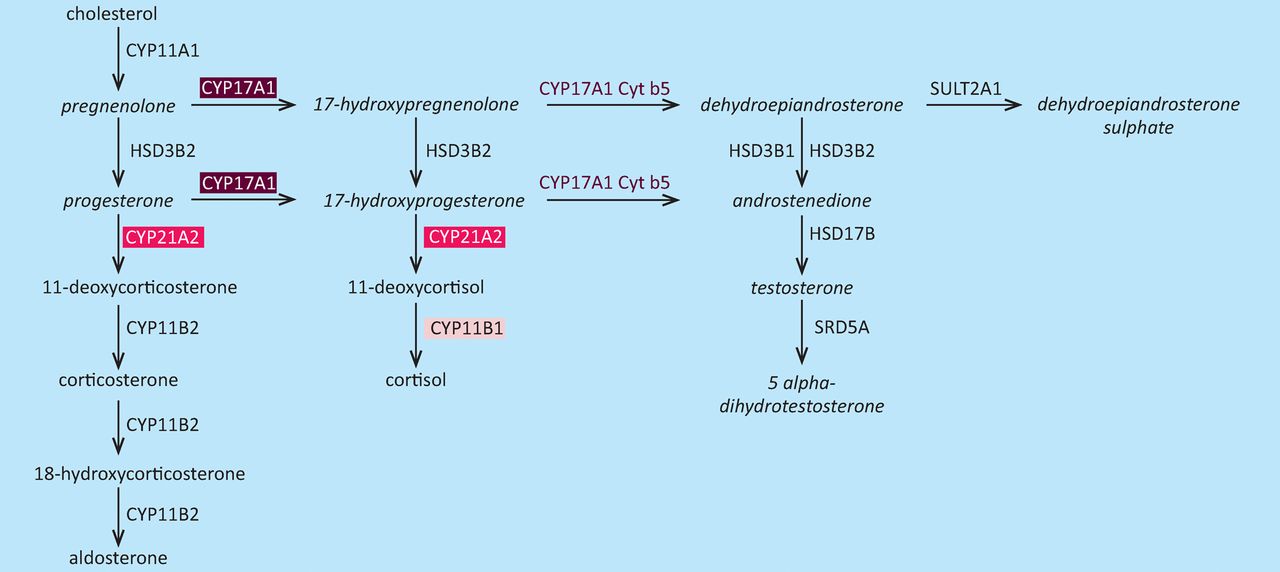
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder characterised by an inborn error of adrenal steroid biosynthesis that results in reduced production of cortisol and adrenal hyperplasia18 (Fig 1).19 Congenital adrenal hyperplasia is due to homozygous or compound heterozygous mutations of the CYP11B1, CYP17A1 and CYP21A2 genes (Table 1), which result in deficiencies of 11β-hydroxylase, 17α-hydroxylase and 21-hydroxylase,18 respectively. Mutational analysis has been used successfully for prenatal diagnosis of CAH when the parents are known to be affected by CAH or to be mutation carriers.20 Children with CAH may present with potentially life-threatening salt-losing crises in the neonatal period and girls with CAH may have virilisation of external genitalia.18 However, early treatment of the pregnant mother of a child with CAH with dexamethasone will prevent virilisation of external genitalia in female foetuses.20 Treatment with dexamethasone is thus started as soon as possible in all at-risk pregnancies to suppress foetal production of androgen, which begins at 6–7 weeks of gestation, and is continued until confirmation that such treatment is not necessary is obtained – that is, until the foetus is confirmed to be a genetic male or an unaffected female. Confirmation by mutational analysis is obtained by chorionic villous sampling at 10–12 weeks of gestation, amniocentesis at 16–18 weeks of gestation or non-invasive prenatal diagnosis of foetal sex, which uses cell-free foetal DNA in maternal plasma, at 7 weeks of gestation.21 Testing of DNA thus has had a major impact on the diagnosis and management of CAH.

Alterations in the steroid biosynthesis pathway in congenital adrenal hyperplasia (CAH). Deficient enzymes are shown in the coloured boxes. Deficiencies in 21-hydroxylase (CYP21A2) (pink box), 17α-hydroxylase (CYP17A1) (maroon box) and 11β-hydroxylase (CYP11B1) (light pink box) result in CAH. In 21-hydroxylase deficiency, which causes about 95% of cases of CAH, production of cortisol and synthesis of aldosterone are reduced and accumulation of precursors such as 17-hydroxyprogesterone leads to increased androgen production, which cause virilisation of female genitalia.18 Steroids produced in excess in 21-hydroxylase deficiency are shown in italics. CAH = congenital adrenal hyperplasia. Adapted with permission from Auchus et al19.
- © 2013 Royal College of Physicians
References








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.