
ABSTRACT
The mechanisms that drive non-alcoholic fatty liver disease (NAFLD) progression from simple steatosis to non-alcoholic steatohepatitis (NASH) and NASH-fibrosis and/or cirrhosis are complex. Recent studies suggest that the liver progenitor cell (ie liver stem cell) population expands during chronic liver injury, and is an essential component of the repair process. Hedgehog (Hh) is a developmental morphogen that has an important role in the adult tissue repair (and progenitor) response. Accumulating data in mice and human show that resurrection of the Hh pathway occurs during progressive NAFLD, and that activity of this pathway correlates with NASH-fibrosis stage. Importantly, Hh ligands secreted by dying (or stressed) hepatocytes, hepatic stellate cells (ie myofibroblasts), cholangiocytes and recruited immune cells can act on neighbouring cells to perpetuate the fibrogenic response. Intriguingly, Hh ligands can also stimulate cholangiocytes to secrete chemokines that recruit immune cell subsets (such as natural killer T cells), which could explain why fibrosis generally occurs in the context of chronic inflammation (ie fibrosis-associated inflammatory response). Finally, the administration of Hh inhibitors led to reduced fibrosis in a model of NASH. Future studies are needed to evaluate the utility of these inhibitors in other models of chronic liver disease. If successful, this could pave the way for the development of new therapy for patients with NASH, because Hh pathway inhibitors have now been licensed for use in patients with advanced basal cell carcinoma.
Background
Non-alcoholic fatty liver disease (NAFLD) is now the leading cause of chronic liver disease and a common indication for liver transplantation among developed countries. The increasing prevalence of NAFLD is related to the tide of obesity, type 2 diabetes mellitus (T2DM) and the lack of physical activity among children and adults. Its close association with putative metabolic risk factors has led to NAFLD being considered the hepatic manifestation of the metabolic syndrome.1,2 NAFLD is, in itself, a risk factor for T2DM, and individuals are also at increased risk of ischaemic heart disease, stroke and certain malignancies, including liver and colorectal cancers. Furthermore, at present, there is no proven therapy for NAFLD. As such, NAFLD is an important public health and economic concern.
NAFLD comprises a spectrum of clinicopathologic states that include simple steatosis (fatty liver (NAFL)), non-alcoholic steatohepatitis (fatty liver with inflammation and hepatocyte death (NASH)) and NASH-fibrosis or cirrhosis (ie degree of scar-tissue accumulation, architectural distortion and organ dysfunction).3 Most patients that are seen in clinics have NAFL, and they rarely develop progressive liver disease (ie fibrosis or cirrhosis). By contrast, NASH is a more ‘advanced’ stage of NAFLD, and a harbinger of fibrosis, cirrhosis and primary liver cancer.4–6 However, only a proportion of individuals with NAFLD will develop progressive disease. Therefore, understanding who, when or how NAFLD progresses will enable clinicians to identify at-risk individuals, and possibly develop new therapeutic strategies.
Pathogenic mechanisms
The mechanisms that lead to the development of NAFL and the subsequent progression to NASH and NASH fibrosis or cirrhosis are complex, and remain to be fully unravelled. Knowledge to date suggests that ectopic, hepatic accumulation of triglycerides (through free fatty acid fluxes), interact with multiple ‘hits’ from cytokines, immune dysregulation and oxidative stress (from mitochondrial dysfunction or endoplasmic reticulum stress) to promote disease progression in genetically susceptible individuals. Recently, changes to the gut microbiota, circadian and epigenetic factors have also been shown to modulate NAFLD outcomes.7–10
In this lecture, I will focus on the final common pathway of NAFLD progression, from NASH to NASH fibrosis or cirrhosis. I will describe the paradigm for liver repair (fibrosis), the repair (fibrosis)-associated inflammatory response paradigm and the role of the Hedgehog (Hh) morphogenic pathway in driving NASH fibrosis.
Model of liver repair
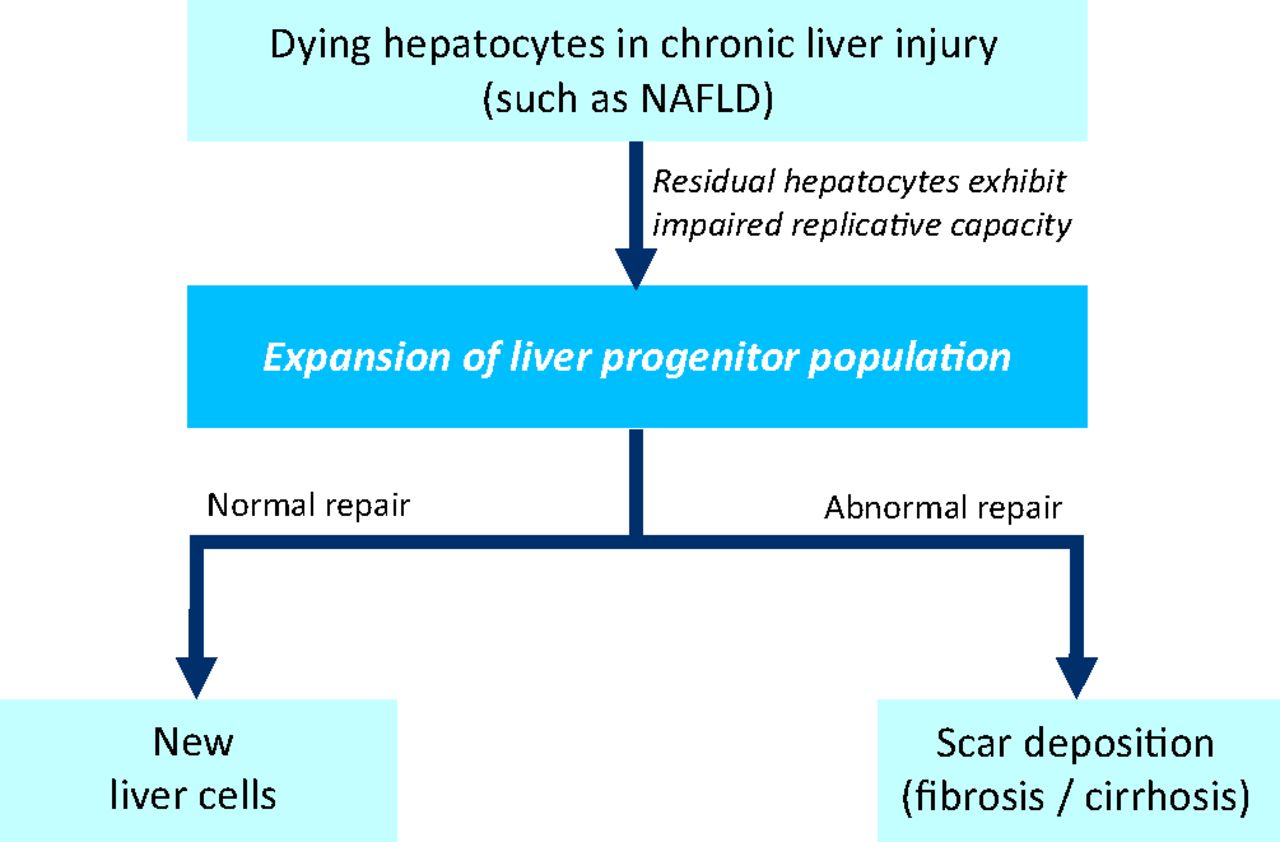
The normal liver has an intrinsic capability to regenerate itself. Liver resection in a healthy liver is followed by replication of remaining hepatocytes to replace the lost (or dying) cells.11 This does not occur during chronic liver disease, because hepatocytes will have been subjected to multiple ‘hits’ as described above (ie oxidative stress, lipotoxicity from free fatty acids, and immune or cytokine stresses), and will have undergone replicative senescence.12 Therefore, during chronic liver disease, the responsibility of liver repair (ie chronic injury response) and attempt at restoration of liver function falls upon the liver progenitor cells (ie liver stem cells) (Fig 1). Liver progenitor cells can be easily identified by immunohistochemistry in paraffin-embedded liver sections from patients with chronic liver disease, using specific liver progenitor markers, such as keratin 7, CD44, epithelial cell adhesion molecule (EpCAM), sex-determining region Y-box 9 (Sox-9) or leucine-rich repeat-containing G protein-coupled receptor 5 (Lgr5). As yet poorly defined factors trigger the expansion and differentiation of liver progenitor cells into new hepatocytes and new cholangiocytes (Fig 1).
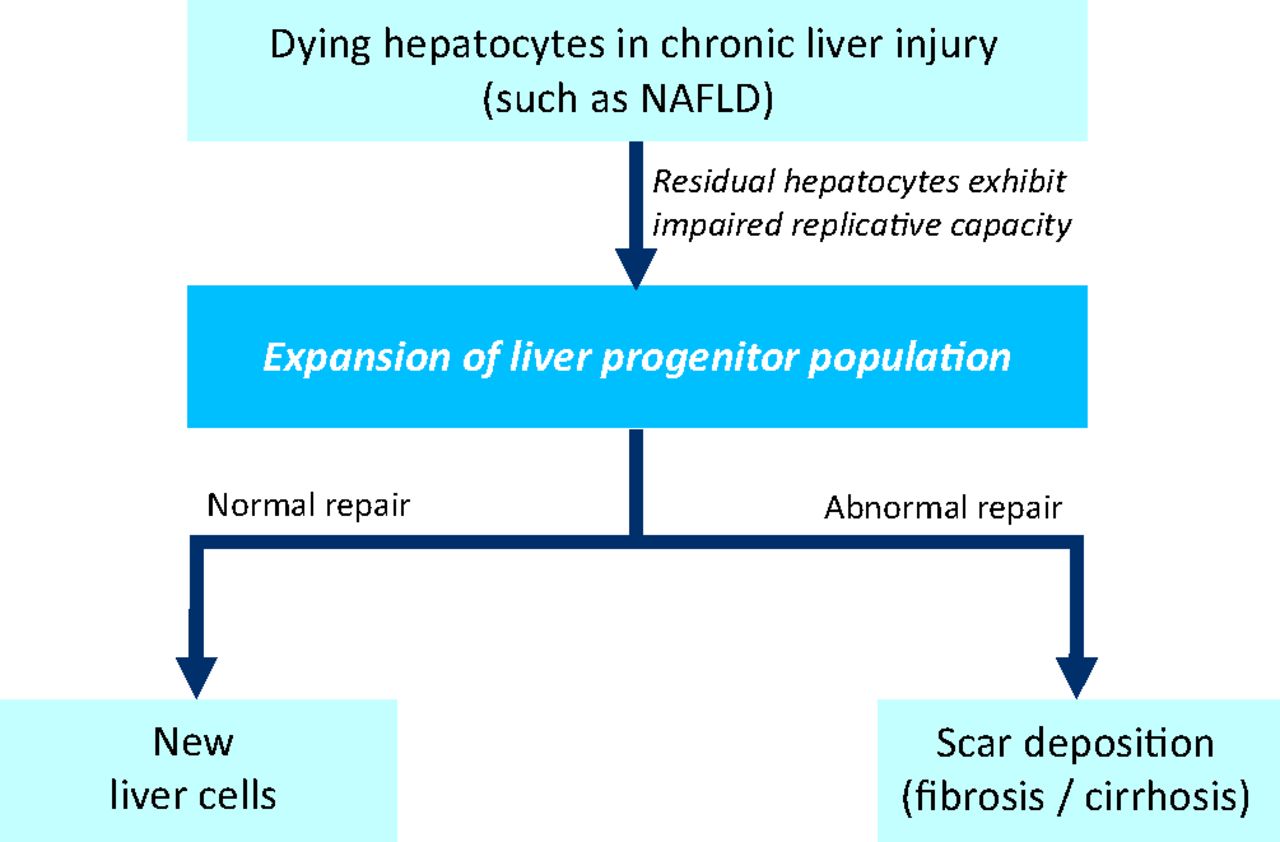
A model of liver repair. During chronic liver injury, such as non-alcoholic fatty liver disease (NAFLD), alcoholic liver disease and viral hepatitis, hepatocytes are subject to ‘hits’ from oxidative stress and immune dysregulation. Hepatocytes exhibit replicative senescence, and are unable to regenerate in response to injury. Instead, the liver progenitor population is triggered to expand, and tasked to replace dying cells and restore hepatic function. Under appropriate conditions, liver progenitors differentiate into new epithelial cells (hepatocytes and biliary cells). By contrast, when exposed to pathological cues (such as excessive or sustained injury, failure of appropriate differentiation, or inadequate protective mechanisms), liver progenitors can directly (via reprogramming) or indirectly (via cytokines or mitogens secreted) promote matrix accumulation (scar formation or fibrosis). NAFLD = nonalcoholic fatty liver disease.
Under pathological cues (as occurring in patients who develop fibrosis or cirrhosis), it is likely that a proportion of liver progenitor cells is ‘reprogrammed’ into a mesenchymal or stromal cell phenotype, which might contribute to scar-tissue (ie myofibroblast) accumulation13 (Fig 1). Recent research also suggests that liver progenitors directly give rise to primary liver cancers.14 The mechanisms that dictate the fate of liver progenitors in adult tissues remain ill-defined or understood, but developmental signaling pathways (such as Notch, Wnt and transforming growth factor (TGF)-β signaling) are likely to be involved.
Role of Hh signaling in liver repair (fibrogenic repair)
Chronic liver injury invokes a wound-healing repair response that resembles tissue construction during development. The role of liver progenitors during adult liver repair led researchers to focus on another developmental molecule, Hh. The Hh signaling pathway was first identified in Drosophilia15,16 and is a highly conserved signaling pathway that orchestrates embryogenesis and tissue remodeling in various systems. It is an important viability factor for stem cells outside of the liver, and deficiency of Hh leads to birth defects, whereas excess Hh activity promotes tumour development. Intriguingly, resurrection of the Hh pathway occurs during repair of adult organs, such as the skin, kidney, pancreas and lung,17–20 and excessive activity of the Hh pathway leads to fibrosis (fibrogenic repair).
Hh pathway activity (expression of Hh ligands and Hh targets) is barely detectable in a healthy liver, but activation of this pathway occurs in mice and human livers after injury. A healthy hepatocyte does not produce or transduce Hh signals. However, when exposed to apoptotic stimuli, mature hepatocytes secrete Hh ligands (Sonic hedgehog and Indian hedgehog) that act on surrounding Hh-responsive cells. These cells include the hepatic stellate cells (resident liver fibroblasts), hepatic myofibroblasts, liver progenitor cells, cholangiocytes, sinusoidal endothelial cells and immune cells.21 In response to Hh ligands, hepatic stellate cells become activated and transition into a myofibroblastic phenotype responsible for matrix deposition (ie scar tissue formation). Activation of the Hh pathway enhances the viability and proliferative capacity of cells, inhibits apoptotic signals and stimulates additional, endogenous production of Hh ligands that perpetuate Hh signaling in an autocrine–paracrine-positive feedback loop.22 Increased expression of Hh ligands also leads to an expanded liver progenitor population, and might promote the ‘reprogramming’ of liver progenitors from an epithelial to mesenchymal phenotype (ie epithelial to mesenchymal transition (EMT)) that is resistant to apoptotic signals, and contributes to scar-tissue accumulation.23 Despite initial controversies, a growing body of evidence shows that EMT occurs as part of repair following chronic injury in adult tissues, and represents the plasticity of cells in response to environmental cues. Similarly, exposure of liver sinusoidal endothelium to Hh ligands leads to an activated phenotype, with loss of fenestration and sieve plates, and increased expression of the activation marker, CD31.24 The aggregate changes described are consistently seen in human liver tissues (ie liver biopsy or explanted liver tissue) as part of the liver repair response during chronic injury (ie fibrosing liver disease). The proximity of Hh-producing and Hh-responsive cells demonstrates a well-orchestrated repair response that is mediated, at least in part, by the Hh pathway.
Hh and immune crosstalk
During scar development, apoptotic hepatocytes, collagen-producing myofibroblasts, liver progenitors and cholangiocytes intermingle with immune cells within fibrous tissues. It was reported that Hh ligands (secreted by neighbouring myofibroblasts) can stimulate cholangiocytes to secrete chemokines (ie chemotactic cytokines), such as CCL2, CCL20, chemokine (C-X-C motif) (CXC)-L11 and CXCL16,25 that chemoattract, retain and activate hepatic stellate cells and immune cells (such as dendritic cells, macrophages, monocytes, neutrophils and lymphocytes) to the injured tissue. For the first time, this provides an explanation for why fibrosis occurs in the context of chronic inflammation; therefore, this was coined the ‘repair (fibrosis)-associated inflammatory response’ (Fig 2).

Repair (fibrosis)-associated inflammatory response. During chronic liver injury, Hedgehog (Hh) pathway activation occurs. Hh ligands (such as Sonic and Indian Hh) are secreted by dying hepatocytes, (myo-) fibroblasts, cholangiocytes and immune cells. These act in an autocrine and paracrine manner to enhance Hh ligand secretion and activate the Hh pathway. Hh ligands stimulate cholangiocytes to secrete chemokines (chemotactic cytokines), such as chemokine (C-X-C motif) (CXC) L16 and CXCL11, which recruit, attract and retain immune cells into the microenvironment (ie chronic inflammatory response).
In addition to promoting inflammatory cell accumulation within the liver, Hh ligands can directly modulate the immune cell phenotype. For example, the Hh ligand, Sonic Hh enhances proliferation, inhibits apoptosis and induces activation of natural killer T (NKT) cells,26 a subpopulation of lymphocytes that often accumulates during liver fibrosis. Furthermore, Hh activation leads to the expansion of a T helper type 2 (Th2) cytokine (such as interleukin (IL)-13)-producing NKT population, thereby providing a pro-fibrogenic microenvironment that helps maintain myofibroblast numbers in the injured liver. These observations are unsurprising because Hh signaling is crucial for thymic progenitor and immune cell development and differentiation. Intriguingly, activated NKT cells can also secrete Hh ligands, and inhibiting the NKT-associated Hh ligands can abrogate fibrogenic outcomes in hepatic stellate cells.27
These findings show that, during chronic liver injury, activation of the Hh pathway modulates the inflammatory response as part of repair.
Hh signaling and repair-associated inflammation in NAFLD
Individuals with NAFL rarely develop significant disease, whereas those with NASH are at risk of developing fibrosis and cirrhosis. The major distinction between NAFL and NASH is the presence of hepatocyte cell death (apoptosis). Using a specific mouse model that exhibits increased hepatocyte apoptotic cell death (the Ikk-β deficient mouse), it was shown that these mice developed greater NASH fibrosis compared with littermates (control) mice when fed a NASH-inducing diet.28 Importantly, the increased amount of fibrosis observed was associated with larger quantities of Hh ligands (Sonic and Indian Hh) produced by apoptotic hepatocytes.
Progressive NAFLD is characterized by a striking liver progenitor response (in mice) or ductular reaction (in humans) that is regulated by the Hh pathway. Neutralising Hh pathway activity using cyclopamine (a Hh pathway antagonist) in mice leads to an attenuated progenitor response and inhibits EMT (see above).29 In turn, this leads to reduced NASH fibrosis. Conversely, mice genetically engineered to exhibit excessive Hh pathway activation develop an exaggerated liver progenitor response, EMT and more fibrosis after diet-induced NASH.
Steatohepatitis, such as NASH, is also characterised by the accumulation of inflammatory cells within the liver (compared with NAFL), and the type and number of inflammatory infiltrate determine NASH outcome. For example, NAFL in mice and humans is associated with depletion of liver NKT cells, whereas NASH is associated with liver NKT cell recruitment and accumulation. Using animal models, it was shown that activation of the Hh pathway directly upregulates expression of key chemokines (such as CXCR3 ligands and CXCL16) that lead to NKT cell recruitment into the liver. Furthermore, mice with an overly active Hh pathway express more CXCL16, and accumulate more liver NKT cells.30 Consistent with earlier observations that immune cells are capable of secreting Hh ligands, it was found that mice genetically deficient in NKT cells (ie CD1d and Jα18 knockout) secrete fewer Hh ligands during injury, and were protected from NASH fibrosis.27 Studies of humans with NAFLD confirm that Hh pathway activation correlates with the severity and stage of NAFLD.31 Furthermore, flow cytometry of liver-derived lymphocytes obtained from explanted NASH-cirrhotic livers confirms that there are significantly more NKT cells in NASH cirrhosis than in normal liver.30
Recently, it was demonstrated that some of the Hh effects might be mediated by downstream effector molecules, such as the matrix molecule, osteopontin (OPN). OPN is expressed and secreted by resident liver cells, such as fibroblasts, cholangiocytes (and liver progenitor cells), and immune cells (such as macrophages, dendritic cells and lymphocytes), and acts in a paracrine and autocrine manner to enhance fibrogenic liver repair, properties that resemble Hh signaling.
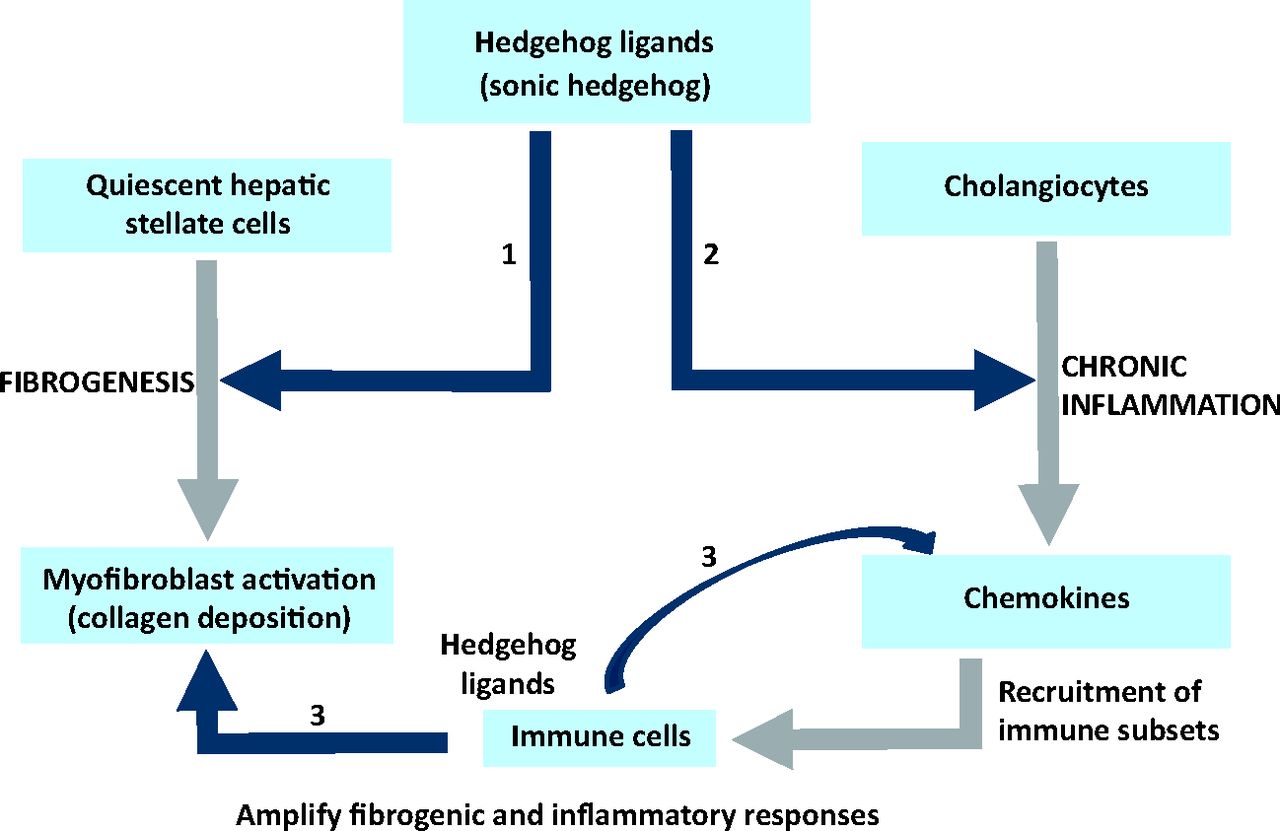
The collective data suggest that a dysregulated, Hh repair response promotes fibrotic outcomes in NASH through multiple, concerted pathways (Fig 3).
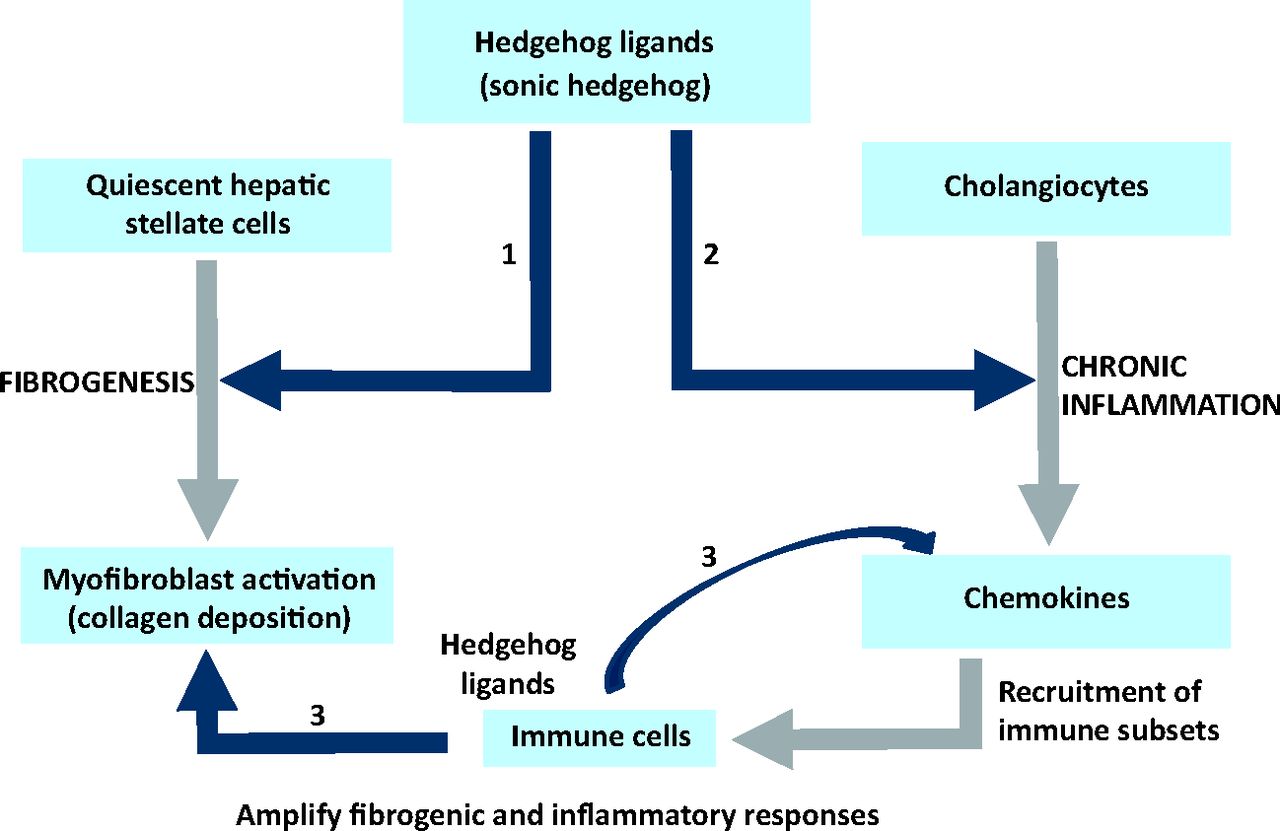
Hedgehog pathway activation promotes fibrogenic outcomes in non-alcoholic steatohepatitis. Hedgehog (Hh) ligands (such as Sonic Hh) secreted during liver injury activate hepatic stellate cells (liver fibroblasts), induce their proliferation and promote transition into a myofibroblastic phenotype responsible for collagen deposition (Pathway 1). Hh is also responsible for the inflammatory response (Pathway 2) during chronic liver disease (also see Fig 2). Recruited immune cells, such as natural killer T (NKT) cells, T cells and macrophages (Kupffer cells), secrete Hh ligands (and other factors) to perpetuate and amplify the inflammatory and fibrogenic responses (Pathways 2 and 3).
Future studies and potential clinical impact
The liver repair response is a conserved, coordinated, wound-healing response that is common to all chronic liver diseases and involves various liver cell types and mechanisms.22 This body of research has thrown light on the role of morphogens, such as Hh, in the adult liver repair (fibrosis) response, and provides compelling evidence for the potential role for Hh inhibitors in patients with advanced NASH fibrosis and cirrhosis. Hh pathway activation also occurs during chronic viral, biliary and chemical-induced liver injury and the degree of Hh pathway activity appears to correlate with disease severity and predict clinical outcomes.
Future studies will be necessary to evaluate the utility of Hh pathway inhibitors in other preclinical models of liver disease. If successful, this could pave the way for the development of new therapies for patients with NASH, because Hh pathway inhibitors have now been licensed for use in patients with advanced basal cell carcinoma.
Acknowledgements
Professor Anna Mae Diehl, Chief of Gastroenterology, Duke University.
- © 2013 Royal College of Physicians
References








{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.