
ABSTRACT
Joint hypermobility syndrome is a common clinical entity which is much misunderstood, overlooked, misdiagnosed and mistreated. It was first described in the 1960s as a purely musculoskeletal condition due to joint laxity and hypermobility occurring in otherwise healthy individuals. Some four decades later it is now perceived to be a multi-systemic heritable disorder of connective tissue with manifestations occurring far beyond the confines of the locomotor system and with ramifications potentially affecting most, if not all, of the bodily systems in one way or another. Most authorities in the field find it clinically indistinguishable from the Ehlers-Danlos syndrome – hypermobility type (formerly, EDS type III). In >50% of patients the diagnosis is delayed for ≥10 years. Failure to diagnose and treat the condition correctly results in needless pain and suffering and in many patients to a progressive decline in their quality of life and in some to a loss of independence.
- Joint hypermobility syndrome
- Ehlers-Danlos syndrome - hypermobility type
- heritable disorders of connective tissue
Introduction
There is a common tendency in medicine when faced with medically unexplained symptoms to assume that they are psychogenic in origin. This is a high-risk approach that can have disastrous consequences. The joint hypermobility syndrome (JHS) story is a good illustration of this.
This is the story of two diseases which, by coincidence, were first both described within a year or two of one another on opposite sides of the river Thames in London. In both conditions, joint hypermobility was identified as a prominent feature. The hypermobility syndrome (HMS) was described by Julian Kirk, Barbara Ansell and Eric Bywaters, rheumatologists at the Hammersmith Hospital in 1967;1 the Ehlers-Danlos syndrome type III (EDS III) was described the following year by Peter Beighton, a clinical geneticist, working a few miles away at St Thomas' Hospital. Beighton had been a former fellow of the late Victor McKusick, the founder of modern medical genetics at Johns Hopkins Hospital in Baltimore.2 He later gave his name to a nine-point scoring system for joint hypermobility, which became universally adopted (by both rheumatologists and geneticists) as a screening test for hypermobility. HMS was regarded (by rheumatologists) as a purely rheumatological disorder in otherwise healthy individuals who happened to be at the upper end of a spectrum of normal joint mobility. EDS III was regarded (by clinical geneticists) as a multisystem heritable disorder of connective tissue (HDCT), with clinical expression in many bodily systems. Unfortunately, despite the geographical proximity of their origins, the two diseases remained separate, each being virtually unknown to the other specialty. This came about for the simple reason that there was virtually no contact (and hence no cross-fertilisation of ideas) between rheumatologists and clinical geneticists. The original concept of each became cemented into the thinking of the 1960s and 1970s and beyond.
With the passage of time and the advent of new knowledge, it has become clear to clinicians seeing many of these patients that, to all intents and purposes, the two diseases are indistinguishable from one another.3 The tragedy is that it took four decades to achieve this consensus. Sadly, even today, there are sceptics who do not yet accept this hypothesis and fervently advocate the separation of the two conditions. For them (principally, but not exclusively, rheumatologists), lax-jointed patients with HMS are ‘otherwise healthy subjects’ whose extra-articular symptoms are taken to represent illness behaviour, rather than bona fide symptoms of systemic complications of EDS III.
One of the principal elements in the argument is the lack of a genetic or other biological marker that can confirm or refute the diagnosis of either of these two conditions. Hopefully, with the current intense focus of research activity in many centres across the world, this crucially important omission will be rectified in the near future. In the meantime, we can only work with what we have, namely clinical studies based on careful clinical observation and evidence based on controlled studies. These studies formed the basis of research conducted in the latter half of the 20th century, which led to greater definition and delineation of the two phenotypes. It is still ongoing.
The realisation that the joint hypermobility syndrome (JHS), as HMS is now principally known, is a multi-systemic illness rather than a mild (seemingly trivial) mechanical disorder of lax joints in otherwise healthy subjects was slow to dawn and even slower to consolidate. When the evidence came, it did so from multiple and sometimes unlikely sources. It is a process that is still unravelling as it becomes increasingly clear to hypermobility-watchers that there is hardly a single medical specialty that is untouched by the condition.4 The evolution of the phenotype over time is illustrated in Fig 1.
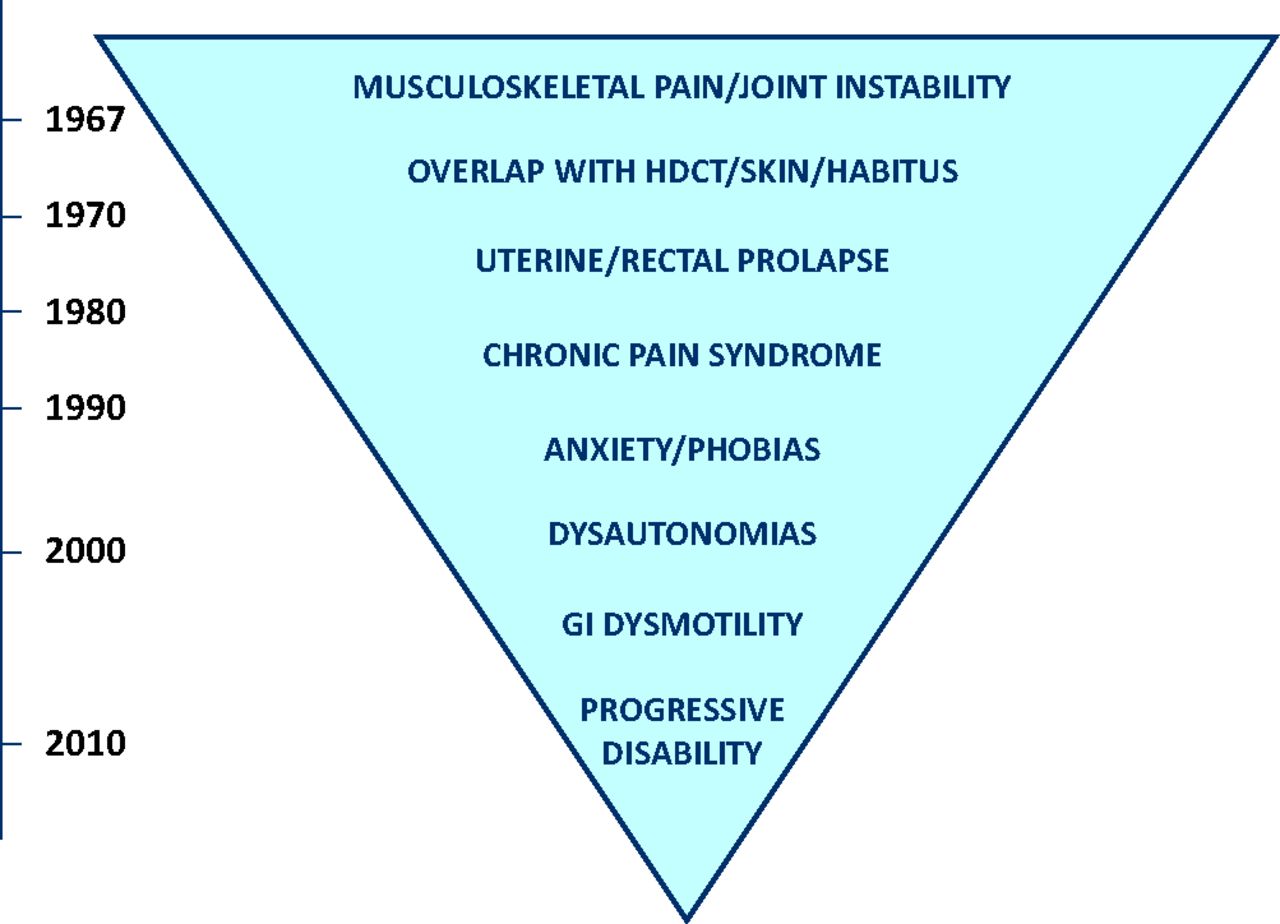
The expansion of the known phenotype during the second half of the 20th century. GI = gastrointestinal; HDCT = heritable disorder of connective tissue.
Thus far, seven key elements, unearthed over the course of nearly half a century, have served to transform our understanding of the nature of JHS, and thereby shape our current concepts of the condition.
1 The overlap with established HDCTs
Soft, silky (and often semi-transparent) skin with increased skin stretchiness (illustrated in Fig 2) in the phase of taking up slack evokes Ehlers–Danlos syndrome (in its various types).5 Identifying an incomplete marfanoid habitus provides a further invaluable diagnostic pointer to JHS,6 while the presence of osteopenia or osteoporosis emphasises the overlap with osteogenesis imperfecta. From the 1980s, the JHS was seen as a milder forme fruste of a composite HDCT, incorporating many of the elements of the better-established and better-known diseases in this category.
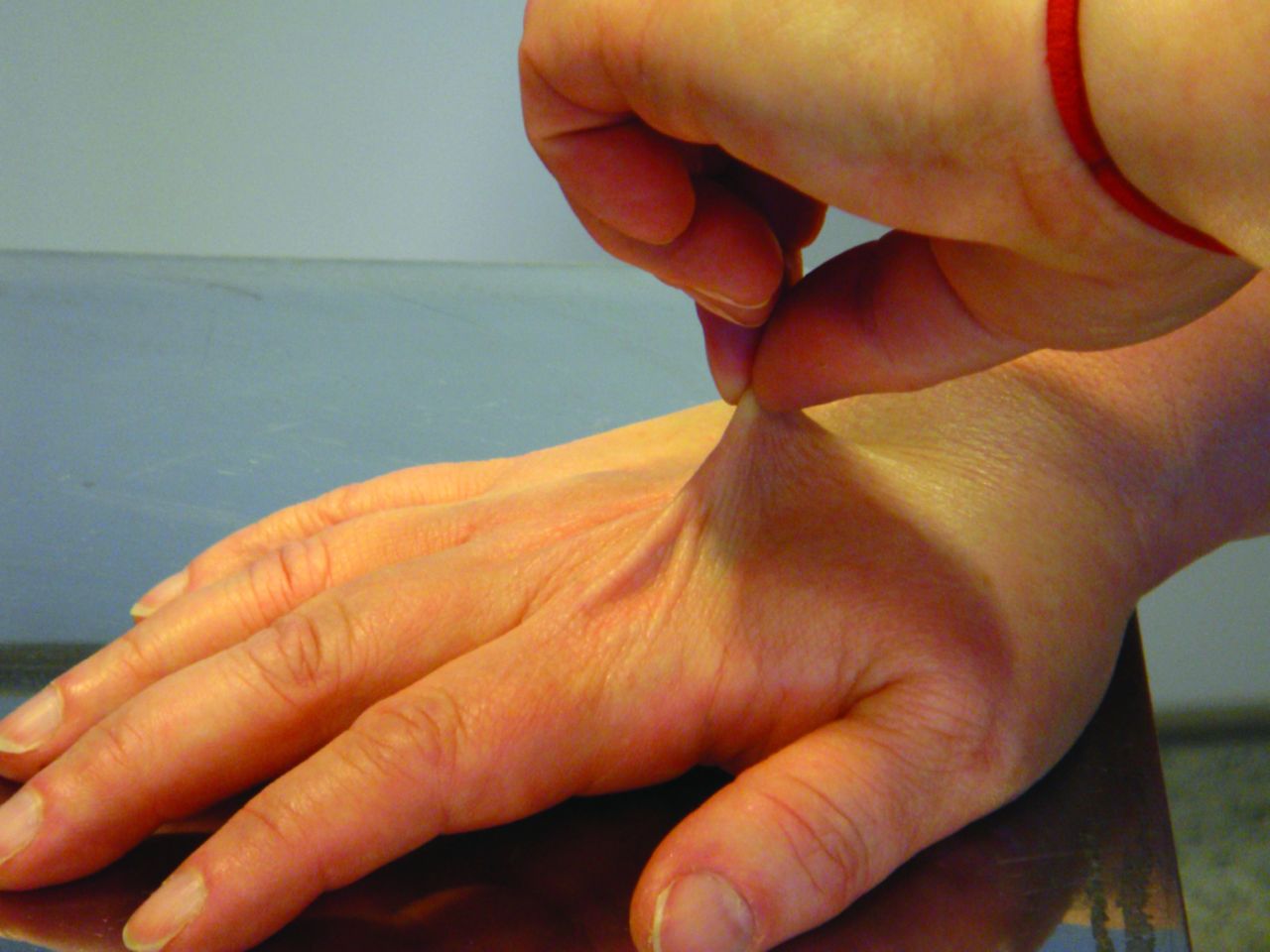
A demonstration of the increased skin stretch seen in JHS. JHS = joint hypermobility syndrome.
2 The important association with uterine and rectal prolapse
The first study to identify an over-representation of hypermobility amongst a cohort of patients with uterine prolapse was published from Iraq as far back as 1982.7 Yet the full detrimental impact that hypermobility can have in women's health is only just emerging.8,9
3 The association with chronic pain
The first clue to the relevance of chronic widespread pain to hypermobility came from the surprising observation that over 50% of all patients admitted to the newly established INPUT Pain Management Unit at St Thomas' Hospital in the early 1990s were hypermobile (V Harding, personal communication). It was some years later that the first study established that intractable chronic pain, which tends to intensify over time, is a frequent occurrence in EDS III.10 Chronic pain in JHS/EDS that is often resistant to all known analgesics has proven to be one of the most difficult complications to treat. The intensity of chronic pain and its impact on the lives of sufferers has been compared with that seen in fibromyalgia and rheumatoid arthritis, and has been found to be greater than that in these other chronic painful diseases.11
4 The relevance of anxiety and phobic disorders
There is no suggestion that psychiatric disorders in general are more highly prevalent in patients with JHS or EDS. Nevertheless, evidence largely emanating from studies in Spain has suggested a firm association between hypermobility and anxiety-related symptoms, in particular panic attacks and certain phobic states such as claustrophobia, agoraphobia and social phobias.12,13 A single report published a decade ago linking these phenomena to a chromosomal duplication (on chromosome 15) has yet to be confirmed in another laboratory.14
5 The high prevalence of dysautonomias
Orthostatic intolerance and other dysautonomic symptoms have been shown to be highly prevalent among patients diagnosed as suffering from JHS, as defined by the 1998 Brighton Criteria.15 In a study conducted among 48 JHS patients and 20 controls, dysautonomic symptoms including pre-syncope, palpitations, chest discomfort, fatigue and heat intolerance were significantly more common among JHS patients than among controls. In this study, 27 patients and 21 controls underwent autonomic evaluation: orthostatic testing and studies of cardiovascular vagal and sympathetic function. Orthostatic hypotension (OH), postural orthostatic tachycardia syndrome (PoTS) and uncategorized orthostatic intolerance (UOI) were found in 78% of JHS patients, compared to 10% of controls. The authors considered dysautonomia to be one of the extra-articular manifestations of JHS.16 A decade later, JHS emerged as one of the most important causes of dysautonomia (especially in PoTS).17
6 The impact of gastrointestinal dysmotility
In the early years of the present century came the additional discovery that the gastrointestinal (GI) tract too may become involved in JHS. Whether this is the result of a collagen defect that affects bowel motility or whether the effect is mediated via an autonomic mechanism is not yet known. What is known is that so-called functional disorders of the GI tract of various kinds are very common. Interestingly, initial studies have shown that the prevalence of hypermobility is surprisingly high among an unselected cohort of patients attending a tertiary referral centre (49% of 129 patients). The full panoply of GI symptoms encountered includes dysphagia, gastro-oesophageal reflux, gastroparesis, slow-transit constipation, pseudo-obstruction, rectal evacuatory dysfunction, rectocele and intussusceptions.18–20
7 The progression to major physical disability – the new ‘rheumatological disability’
As JHS is a genetically determined disorder, the underlying defect, which is likely to be a mutation affecting one of the genes encoding one of the fibrous proteins of the connective tissue matrix (a collagen, elastin, fibrillin or tenascin), is determined at conception. Many people who inherit the defect remain symptom-free throughout their lives, whereas others may develop minimal musculoskeletal symptoms after unaccustomed exercise. The full panoply of complications does not appear all at once, but gradually over the first 2–3 decades of life.
The natural history of JHS can be seen to comprise three principal phases. The first phase results from connective tissue laxity and fragility and comprises multiple soft tissue injuries, joint and spinal instability and dislocation, and complications of weakness in supporting structures (pelvic floor, herniae and varicose veins). Phase 2 is the superimposition of non-articular complications: pain amplification, kinesiophobia deconditioning (widespread chronic pain often described as ‘fibromyalgia) and fatigue (which is often prominent and may be misdiagnosed as CFS/ME). It is in this phase that orthostatic intolerance, PoTS and other dysautonomic features as well as GI symptoms make their presence felt. The final phase is the emergence of psychosocial sequelae such as anxiety or depression, obesity (often associated with comfort eating), work incapacity, isolation and despair. At this stage, there is often a downward spiral of loss of mobility, self-efficacy and self-esteem as the quality of life diminishes. The reasons for this decline are complex and multifactorial. At this stage, the patients are severely disabled in spite of the fact that their musculoskeletal system is usually grossly intact. It would appear that the combination of overriding chronic pain (which is largely unresponsive to analgesics) and systemic components (PoTS and GI problems) is the determining factor in this decline. Treatment for more severely affected patients has, until recently, been non-existent; in recent years, collaborative units have sprung up across London and beyond try to offer patients some prospect of improvement. These units comprise University College London Hospitals (UCLH – clinical assessment and pain management), Royal National Orthopaedic Hospital (RNOH) (intensive rehabilitation with pain management), Bart's Health (gastroenterology), the National Hospital for Neurology & Neurosurgery (autonomic dysfunction), and the Hospital of St John & St Elizabeth (one-stop hypermobility Unit). Initial experience suggests that a combination of physical rehabilitation adapted to the needs of patients with lax and fragile tissues, coupled with pain management using cognitive behavioural techniques, provides the way forward for the treatment of both the JHS and the PoTS. Further evaluation is currently in progress.
Conclusion
Far from being a minor disorder occurring in essentially normal individuals, as is perceived by many of our colleagues even today, there is abundant evidence of a multisystemic hypermobility-related disorder that causes much suffering in the community. This is an emerging disease, which bears little resemblance to the joint hypermobility diseases originally described in the 1960s. To dismiss disparate symptoms as ‘illness behaviour’ is, in my view, wrong and does much disservice both to our patients and to our reputation as a profession. More often than not, JHS is still overlooked, misdiagnosed and hence untreated.21
- © 2013 Royal College of Physicians








{kind=link}
{kind=link}