
ABSTRACT
Acute myeloid leukaemia is a heterogeneous disease that occurs in all age groups but peaks in older age at around a median of 69–70 years where it has a frequency of 13–15/100,00/annum. With the changing demographics, the number of cases will increase in line with the older population. As the only treatment with curative intent is intensive chemotherapy, this presents an immediate therapeutic challenge for the majority of the disease.
Biological insights
An accepted view of normal haematopoiesis is that the various familiar lineages resulting in the cellular components of the blood derive from a population of stem cells that are morphologically indistinct, but have the unique characteristics of being able to self-renew to sustain longevity and, under the influence of appropriate micro-environmental factors, differentiate into specialised blood cells, which thereby progressively lose the ability to proliferate. Leukaemia could be viewed as a failure of differentiation, such that mature progeny are progressively not produced and the more proliferative precursor ‘blast’ cells accumulate. The result is that the diagnosis is not usually difficult to make, because the patients frequently present with the consequences of cytopenia reflected in the peripheral blood and, on marrow examination, an excess of blast cells. Therapeutically, it is important to know whether this problem has originated in the myeloid lineage or the lymphoid lineage because the treatments and therapeutic outcomes are substantially different. This can be resolved diagnostically by associated cytochemistry and/or immunophenotyping.
Under the microscope, there is considerable heterogeneity that is reflected in a historical morphological classification called the ‘FAB’ system. In general, the morphological classification has less use with respect to prognosis because of the associated use of cytogenetics, which although adding modest diagnostic confirmation of some subtypes is of crucial importance to prognosis.1,2 Sixty to 70% of patients will have a non-random cytogenetic lesion, many of which have clear prognostic implications. Of patients aged <60 years, approximately 25% have a lesion that predicts a favourable outcome (~75% survival), approximately 15% have a lesion that predicts a poor response (20% survival), and the remainder have an intermediate prognosis of around 45%. In one case in particular, the chromosome change (a t(15;17)) is associated with characteristic morphology and is now regarded as a separate disease with an expected cure rate of 85%. The challenge in this subgroup is to minimise chemotherapy, and very recent data suggest that this is possible with the combination of the vitamin A derivative all-trans retinoic acid (ATRA) and arsenic trioxide.3 The understanding of why ATRA is so effective did not emerge before it was observed empirically that it worked in vitro. The explanation subsequently emerged that sensitivity is uniquely associated with t(15;17), because a consequence of the translocation was disruption of the sequence that encodes the retinoic acid receptor. Unfortunately, this outstanding sequence of events has not yet been reproduced with other agents in other acute myeloid leukaemia (AML) subgroups. Leukaemia cells do show sensitivity to arsenic in vitro, but attempts to transfer this to clinical benefit have failed.
In addition to the spectrum of non-random cytogenetic abnormalities, many of which have important prognostic implications, it is now clear that there is considerably more heterogeneity at the molecular level whereby 80–90% of patients will have a mutation in one of 15–20 genes.4,5 These also may have independent prognostic implications and, of course, in some cases they are targets for therapy. Many of these abnormalities disrupt cell signalling or cell death pathways that facilitate proliferation or avoidance of apoptosis. More recently, epigenetic lesions have also complicated the picture.6
There is a clinical incentive to use this sort of information as a prognostic guide to direct treatments in individual patients, in particular who should have remission consolidated with a stem cell transplant and who should not. This has already been useful, but remains complex because the molecular abnormalities frequently do not occur alone and the combination can modify the prognostic information. It is likely that over the next few years, as more information accumulates, the situation will become clearer.
Treatment
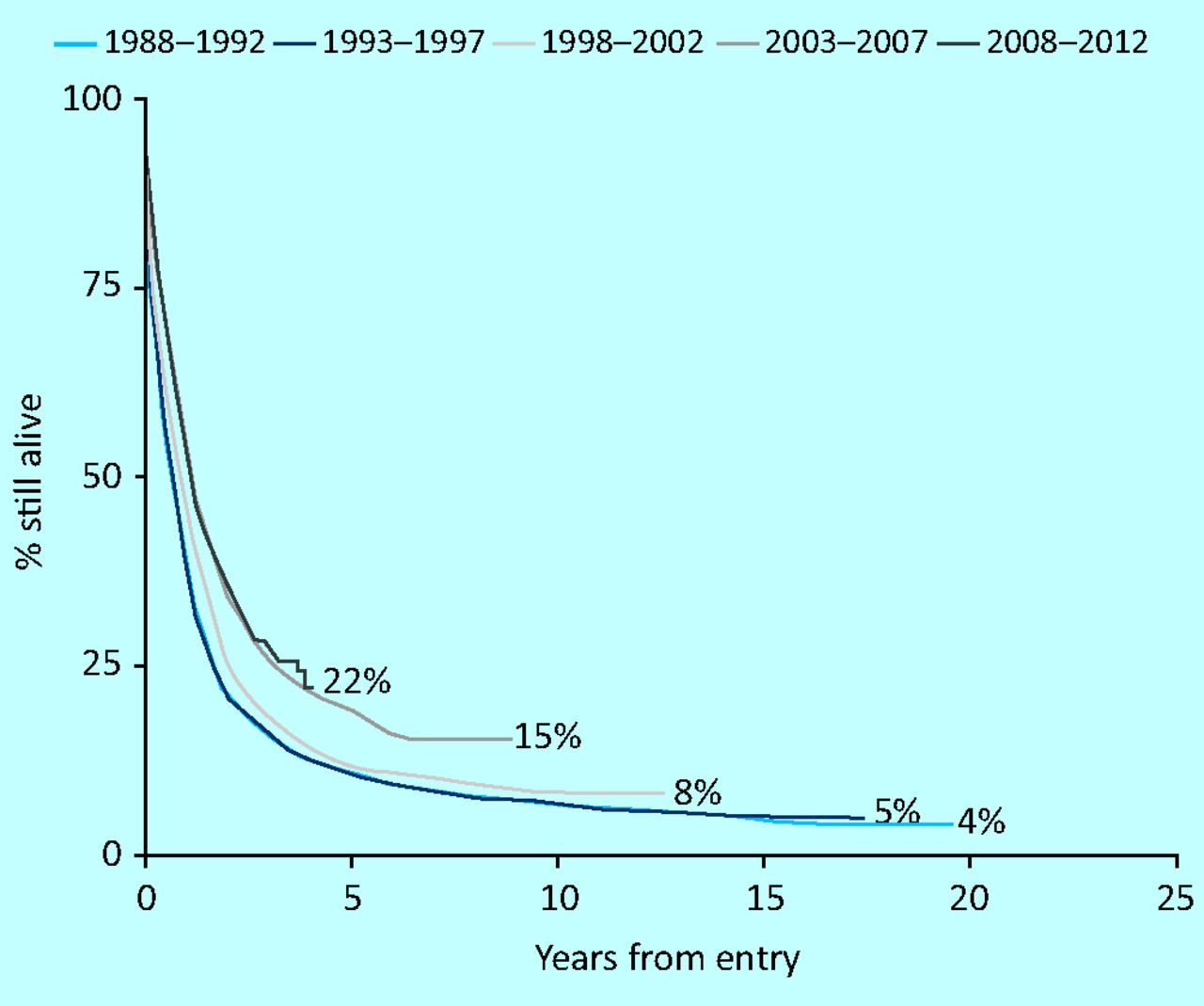
The approach to treatment with chemotherapy has changed little for the past three decades and comprises the combination of a DNA intercalating agent (usually daunorubicin) with a nucleoside analogue (usually cytosine arabinoside [ara-C]).7 Despite the lack of therapeutic innovation there has been a steady improvement in survival in younger patients (Fig 1), but for reasons to be discussed progress in older patients has been disappointing (Fig 2). Many studies have compared alternative intercalating agents, doses of intercalating agents, alternative nucleoside analogues, dose and schedule of nucleoside analogues, or the addition of a third drug, without providing convincing evidence of a fundamental advance. However, this is rigorous treatment and initially will put the patient at significant risk owing to the need to cause transient marrow ablation with the hope that the leukaemic clone will be substantially eliminated while enabling normal haematopoiesis to re-emerge usually around 3–4 weeks later. This ‘selective weedkiller’ approach will result in functional normalisation of marrow function in 75–80% of younger patients (<60 years), but only 50–60% of older patients.8 5–6% of younger patients and 10–12% of older patients will die as a result of complications associated with this treatment (eg infections, bleeding or organ failure). 10–20% will fail due to inability to eradicate disease.

Survival in patients under 60 years in UK trails.

Survival in older patients in UK trials.
The definition of ‘remission’ depends on viewing an adequately cellular marrow smear under the microscope and detecting <5% of the cells as residual leukaemia, but this needs to be accompanied by recovery of the peripheral blood neutrophil and platelet count. The emergence of the molecular information that lends itself to polymerase chain reaction, and more sophisticated characterisation of the leukaemic population at diagnosis, enables a more refined level of detection of 1 in 104 or 105 of residual or re-emerging disease.9 This technology has demonstrated that many patients in morphological remission still have evidence of residual leukaemia at this level and this is associated with a higher risk of relapse. It has long been known that even though remission has been confirmed, further courses of chemotherapy are required, but how many is less clear. Contemporary evidence in the UK indicates that there is no advantage in more than a total of four courses for younger patients and two or three courses in total for older patients.10 Some patients will be given a stem cell transplant as a way of consolidating remission, but this is only applicable to a minority of patients as will be discussed below.
Given that the disease is more common in older patients and demographic factors will result in the disease becoming more frequent in future years, the treatment of older patients has attracted increasing attention. Because the treatment is tough, and older patients accumulate co-morbidities, intensive chemotherapy is more risky. Epidemiological studies suggest that as few as 30% of patients will receive treatment with curative intent.11 This may or may not be appropriate. Retrospective studies of older patients who were selected for conventional chemotherapy indicated that with increasing age and deteriorating performance scores, the risk of treatment failure and early mortality is as high as 80%.12 However, population studies from Sweden suggest that survival is slightly better in geographical regions when an intensive approach is normally given.13 Additional information provided by cytogenetics could inform the choice of treatment because, even for fit older patients, adverse cytogenetic changes indicate a modest chance of useful response. There is no adequately sized trial to compare an intensive vs non-intensive approach by randomisation in older patients. These older frailer patients have a median survival of approximately 3 months if treated palliatively. This justifies the growing number of investigational agents being tested in this population.
Why does treatment fail?
A number of prognostic factors have been independently shown to predict a better or worse prognosis. Rising age, high presenting white cell count, cytogenetic abnormality and whether the disease arises de novo or secondarily to a preceding haematological syndrome are all well known to indicate a poorer response to treatment. Although age is an independent factor, it is partly explained by the accumulation of adverse factors in older age. It is not clear why these factors predict an adverse outcome, but various biochemical mechanisms of drug resistance tend to be more frequent in older patients or in those with adverse cytogenetics. The best characterised is P-glycoprotein, which can efflux drugs such as daunorubicin from the cell. This became a therapeutic target based on convincing pre-clinical evidence that this ‘efflux pump’ could very effectively be blocked by various drugs such as cyclosporin or its derivatives, but it was not effective in clinical trials.
Recent molecular information confirms that genetic subclones, which may or may not have been present at diagnosis, escape chemotherapy and emerge as relapsed disease. Although some of these mutations may not be causative, but rather, ‘passenger’ mutations, their emergence may have implications for molecularly directed treatments that are now entering prospective trials. The same biology is emerging in other cancers.
Incorporation of molecular information
Recent years has been characterised by a proliferation of molecular information indicating that many mutations are associated with the disease. Many of them have positive or negative prognostic value, but it is less clear as to whether they can predict which treatment to apply. Approximately 90% of patients can be found to have a mutation in one of 15 genes;4 however, as increasing information emerges, it is clear that the prognostic reliability is complicated because of the co-existence of more than one mutation in approximately 60% of patients. It is also emerging that not all of the mutations are initiating events, but rather ‘passenger’ events. Nevertheless, there is therapeutic interest in incorporating small molecule inhibitors, which are usually tyrosine kinase inhibitors, into treatment. So far, none has been shown to produce durable responses and it is concluded that the best chance may be to combine them with chemotherapy.
The most frequent mutations are of the Fms-like tyrosine kinase 3 (FLT3) receptor, which occurs in approximately 25% of younger patients and approximately 15% of older patients and does not influence the prospect of achieving remission, but independently predicts a higher risk of relapse.14 However, the predictive value is complicated by the observation that the higher the allelic ratio of mutant to non-mutant alleles, the higher the relapse risk. An FLT3 mutation frequently occurs with a second mutation of nucleophosmin1, which usually functions as a shuttling protein. Approximately 40% of younger patients will have the nucleophosmin1 mutation, but in this case, if occurring alone, it offers a reduced risk of relapse, unless it co-exists with a FLT3 mutation. This type of combination information is emerging regularly.
Stem cell transplantation
The most effective way to prevent disease recurrence is allogeneic transplantation.15 In its early days, it was assumed that this was merely a way of intensifying treatment of the host bone marrow, by using total body irradiation, which was ablative, and using the donor marrow to ‘rescue’ haematopoietic function. However, it was noticed that the same strategy was less successful when used in identical twins. This led to the understanding that the mechanism by which an allogeneic transplant effected cure was immunological and mediated by donor T cells, a so-called ‘graft versus leukaemia effect’. The results were best when there was a close human leukocyte antigen (HLA) match between donor and host, which was usually found within families. However, it has now become clear that a close match between unrelated pairs can now deliver equivalent results. This sort of myeloablative transplant begins to lose its benefit when the patient's age exceeds 35–40 years. Therefore the donor matching and age requirements placed significant limitations on this approach. In recent years, it has become clear that a donor's immune system can be engrafted without the need for conventional myeloablative total body irradiation, thus offering this mechanism for older patients, even up to the eighth decade. Although convincingly demonstrating the feasibility of this reduced intensity conditioning (RIC) approach, comparative studies have been scarce. Recent UK studies suggest that RICs offer older patients who have a matched sibling donor a superior survival, but so far only if they have intermediate and not high-risk disease. Similarly, it is not clear that these sorts of transplant between matched, but unrelated, donors offer an advantage. International donor panels have made unrelated transplants feasible, depending on the ethnic background. The emergence of cord blood cells as an effective source also expands options.
The older frail patient
Given that the median age of patients at diagnosis is 68 years, and older age is associated with co-morbidity, providing adequate treatment for this patient group is a challenge. Not only is the disease biologically more difficult, but also the rigours of therapy present significant risks. Traditionally, it was accepted that conventional therapy was too risky and palliation with transfusion and antibiotic support with drugs such as hydroxycarbamide was the normal approach. A UK study compared this approach with regular low-dose cytarabine (20 mg twice daily for 10 days by subcutaneous injection), and it became clear that this was superior because it was able to deliver 15–20% remissions, but provided little benefit for those who did not achieve remission.16 There was no increase in supportive care requirements or recorded toxicities, so this has become a standard of care. A newer development has been epigenetic therapy in the form of demethylating agents.17,18 These are assumed to work by activating genes, although there is no clear correlation of demethylation with clinical response. These agents are useful for some patients who have a non-proliferative form of AML. They tend not to deliver remissions, but can stabilise disease for a few months. No randomised data has yet convincingly shown either demethyating agent to be superior to low-dose ara-C, as used in the UK trials. Several new agents are being tested in this patient group.
Conclusions
AML is a challenging disease because of extensive morphological, immunological, cytogenetic and molecular heterogeneity. Although progress with chemotherapy has been made, particularly in younger patients, it may be close to reaching its potential. To an extent ‘personalising’ treatment has been ongoing for some time, particularly the choice of stem cell transplant. There is much optimism that molecularly targeted treatments will be the next step forward, but proof of that is awaited. More older patients will develop the disease and that is one of the current areas of therapeutic interest, but this is a difficult problem to solve.
- © 2013 Royal College of Physicians
References








{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.