
Key points
Despite multidisciplinary care, the prognosis for patients with cystic fibrosis (CF) remains limited
Small-molecule treatments aimed at restoring the function of the CF transmembrane conductance regulator (CFTR) are offering promising results for mutation-specific defects
For those with the G551D mutation, ivacaftor is now available in clinical practice
Small-molecule medications are not yet available for all patients with CF
In the future, gene therapy may offer effective treatment of CF irrespective of the genetic defect
Introduction
Cystic fibrosis (CF) is the most common, life-limiting, autosomal-recessive genetic disease. In the UK, it affects about 10,000 people (one in 2,500 live births).1 The -traditional treatments available for patients with CF focus on the consequences of this genetic abnormality. In development are a group of treatments that are focused on overcoming this genetic defect directly or indirectly.
Pathophysiology
The CF gene is found on the long arm of chromosome 7 and encodes for a 1,480-amino-acid protein known as the cystic fibrosis transmembrane conductance regulator (CFTR).2 More than 1,900 mutations of the CF gene are now recognised, of which F508del (a deletion of the amino acid phenylalanine at position 508) is the most common.3–4
The CFTR protein is trafficked through the cell via the endoplasmic reticulum and Golgi apparatus, where it is inserted into the apical membrane of epithelial cells and functions as a cyclic adenosine monophosphate (AMP)-dependent chloride channel (Fig. 1).5 Its pathophysiological effect in CF is reduced chloride conductance at epithelial membranes, which results in secretions with abnormal chloride and sodium concentrations. The clinical consequences of absent or reduced CFTR function are the complex, multi-organ characteristics of the CF phenotype. This results in excessively viscous secretions in the lungs and excess secretion of sodium chloride in the skin (which is the basis for the sweat test used in diagnosis of CF).
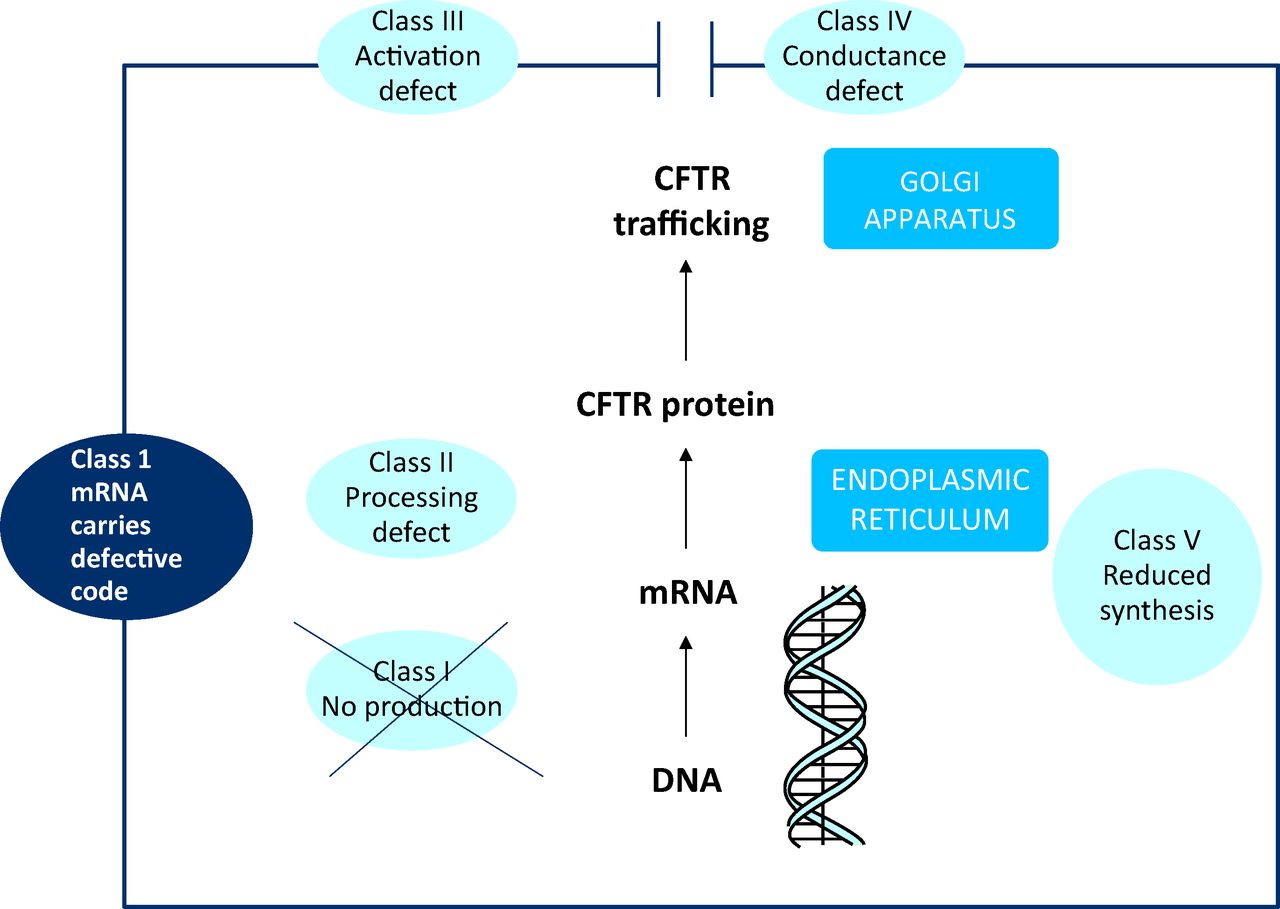
The cystic fibrosis (CF) gene encodes for a protein, CF transmembrane conductance regulator (CFTR), which is trafficked through the cell and inserted into the apical membrane, where it functions as a chloride channel. CF = cystic fibrosis; CFTR = cystic fibrosis transmembrane conductance regulator; DNA = deoxyribonucleic aicid; mRNA = messenger ribonucleic acid.
Mutations in the gene encoding the protein CFTR can be divided into five categories on the basis of the effect they have on protein synthesis:6
Class I defects (eg G542X) disrupt synthesis of CFTR and include nonsense and frameshift mutations that lead to premature termination codons (PTCs) and a lack of protein production.
Class II defects (eg F508del) result in misfolded CFTR that is then degraded in the endoplasmic reticulum.
Class III defects (eg G551D) result in CFTR that reaches the apical membrane but is not activated and is therefore non-functional.
Class IV defects (eg R117H) result in reduced conductance of CFTR at the cell surface.
Class V defects (eg A455E) result in overall reduced synthesis of normal CFTR.
Class I–III defects result in no CFTR function at the cell membrane and are associated with more severe phenotypes. In patients with class IV and V mutations, the CFTR protein may have some degree of function, which results in a less severe -phenotype.7
Multidisciplinary approach
Improvements in the organisation and delivery of care to patients with CF have led to modest benefits for individual patients. However, their cumulative effects have improved quality of life and prognosis for patients living with CF (Box 1). Although survival has improved substantially over the past 20 years, the median age at death remains 27 years.1
Box 1 Pillars of modern management of cystic fibrosis.

The start of a new era
The traditional treatments available for patients with CF have focused on the consequences of abnormal CFTR function. For the first time, there are now treatments designed to overcome specific mutation abnormalities that restore the function of the defective CFTR protein.
Small-molecule CFTR modulators
This promising class of drugs can be divided into three therapeutic categories:
agents that promote ribosomal readthrough of nonsense mutations (eg ataluren)
’CFTR correctors’ (eg lumacaftor)
’CFTR potentiators’ (eg ivacaftor).
Class I defects
Class I defects account for up to 10% of all CF mutations and more than 60% in selected population groups (eg Israeli patients).8 These nonsense mutations result in the insertion of a PTC in transcribed messenger ribonucleic acid (mRNA). The CFTR protein produced is usually truncated and non-functional, which results in a typically severe phenotype of CF.
Ataluren is a small-molecule drug designed to make ribosomes less sensitive to PTCs, which allows a ‘readthrough’ and the production of functional CFTR protein. The results of a recently conducted phase-3, randomised, double-blind placebo-controlled clinical trial showed a 3% improvement in lung function (measured as Forced Expiratory Volume in 1 second (FEV1) at 48 weeks) in the Ataluren arm. This result did not, however, reach statistical significance. The secondary endpoints were more promising, demonstrating a 23% reduction in rate of protocol-defined pulmonary exacerbations (p value = 0.0992).9 Aminoglycosides have been shown to bind the same site in ribosomes as ataluren, thus negating its effect. When patients taking aminoglycoside antibiotics were excluded from the analysis, significant treatment effects with ataluren were seen in terms of both FEV1 (6.7% compared to placebo) and exacerbation rates (pulmonary exacerbations decreased by 43% in treatment group).
Class II defects
The class II mutation F508del accounts for two-thirds of all CF mutations worldwide, and about 50% of affected patients are homozygous for this mutation. The F508del mutation disrupts normal folding and trafficking of CFTR through the cell, which results in little to no expression of CFTR at the epithelial surface.
The CFTR ‘correctors’, such as lumacaftor, improve trafficking of CFTR through the cell, which increases the quantity of CFTR expressed at the cell surface. When used in combination with a CFTR ‘potentiator’ such as ivacaftor, it is hoped that meaningful CFTR function can be achieved.
A programme of phase II studies has assessed the safety and efficacy of lumacaftor alone and in conjunction with ivacaftor in subjects homozygous for the F508del mutation (NCT 01531673). No significant effects were noted from single-agent treatment with lumacaftor. However, 28 days of combination treatment with lumacaftor and ivacaftor led to statistically and clinically significant improvements in FEV1 of about 6% (p < 0.01 vs placebo).10 Based on these findings, two phase III confirmatory studies, TRAFFIC (clinicaltrials.gov identifier: NCT01807923) and TRANSPORT (NCT01807949), have been initiated to evaluate the effects of lumacaftor in combination with ivacaftor in patients homozygous for the F508del -mutation.
Another CFTR ‘corrector’ in development is currently known as VX-661. Preliminary results from a phase II study of its use in combination with ivacaftor have shown promising results in patients homozygous for the F508del mutation (NCT 00909532).11 It is likely that a phase III programme will be developed shortly.
Class III defects
The class III mutation G551D accounts for about 5% of all CF mutations. Ivacaftor ‘potentiates’ CFTR in patients with the G551D mutation, increasing the amount of time the chloride channel remains open and so improving CTFR's function at the epithelial surface.12
A randomised, double-blind, placebo-controlled clinical trial in more than 160 patients confirmed its fundamental effect on the CFTR protein, with a mean decrease in the concentration of chloride in sweat of 48 mmol/l (often to levels <60 mmol/l, such that they could be regarded as normal), which was sustained over a 48-week period. This translated into improvements in lung function (improvement in FEV1 of more than 10 percentage points), exacerbation frequency (overall reduction of 55%) and quality of life. Its effect was seen across the spectrum of disease severity.13
Ivacaftor was first made available for clinical use in the UK in 2013 via specialist commissioning. Comparable improvements in lung function and exacerbation rates are being widely reported in clinical practice.
Class IV defects
Ivacaftor is also being studied in patients with the class IV defect R117H. The KONDUCT study (NCT01614457), which has the primary endpoint of change in FEV1 over a 24-week period, is aiming to enrol 60 patients.
Gene therapy
In contrast to mutation-specific, small-molecule therapy, gene therapy attempts to introduce a normal functioning copy of the CFTR gene into the cells of the conducting airways. If successful, this approach will be available to all patients with CF irrespective of the underlying genetic defect.
More than a decade of investigation has led to the development of a product that offers the best currently available -gene-transfer potential. A combination of a CFTR-expressing plasmid (pGM169) with a cationic lipid (GL67A) delivered via nebuliser has been assessed in phase I and IIA safety studies (Eudra CT ref 2007-004060-85; NCT 01621867).14 A phase IIB, double-blind, placebo-controlled, parallel-group, multi-dose study (NCT01621867) has recently completed recruitment, with results expected in 2014.
Conclusion
For the first time, treatments that target the underlying defect in CF – improving the function of the CFTR protein – are available. Within the next decade, it is envisaged that it will be routine to deliver individualised therapy designed to overcome a -specific mutational abnormality. It is hoped that this will have a fundamental impact on the course of the disease, significantly improving quality of life and prognosis.
- © 2014 Royal College of Physicians








{kind=link}