
Since 2007 genome-wide association studies (GWASs) have identified hundreds of common genetic variants (usually single nucleotide polymorphisms [SNPs]) associated with common diseases and traits. This information is catalogued online (see www.ebi.ac.uk/fgpt/gwas for an interactive diagram). To date, there are few examples where GWAS findings have translated into useful tests that can help individual patients. Instead the common variants identified by GWASs have provided a much-needed first step in improving understanding of common disease aetiology. In this short review, I discuss some of the advantages of GWAS approaches, including the biological insights arising from GWAS, and some of the limitations. Examples I focus on include type 2 diabetes and related metabolic diseases.
What is a genome-wide association study?
GWASs typically involve the direct genotyping of several hundred thousand SNPs in hundreds to thousands of DNA samples using microarray technology. After stringent quality control procedures, each variant is analysed against the trait of interest. Typically researchers will collaborate and combine data from studies with the same disease or trait available. As more studies have performed genome-wide genotyping in the last 6–7 years, GWASs using individuals of European ancestry have reached, for example, more than 250,000 individuals for height and BMI,1,2 32,000 Europeans for type 2 diabetes,3 and 190,000 individuals for lipid levels.4 Similar GWAS efforts have been performed in other major ethnic groups, eg for type 2 diabetes; the latest studies include 7,000 east Asian,5 5,500 south Asian,6 and most recently 3,800 Latin American7 and 6,000 Japanese8 type 2 diabetes cases.
In contrast to the candidate gene and linkage study era before 2007, where many findings in common disease genetics proved to be false positives, the vast majority of associations identified by GWASs are extremely robust statistically and are reproducible in additional studies. Most findings have been reproduced in different ethnic groups, with some differences in allele frequency, eg the variants identified as associated with lipid levels in Europeans are also associated with lipid levels in Asians and, to a lesser extent, Africans.9 In contrast to the linkage-based approaches, such as the use of affected sib pairs, GWAS findings identify small, tractable regions of the genome where, for most loci, the likely target gene is one of only a small handful of genes – typically 2–4, although some loci may contain 0, and some 20, genes. Most of these variants lie at the opposite end of the frequency and penetrance spectrum compared with mutations that cause monogenic diseases. Typically the frequency of a risk allele is >5%, meaning that more than 1 in 10 people will often carry the disease risk allele. As even common diseases typically affect only 1–5% of the population, the vast majority of risk allele carriers are unaffected and, because common diseases are polygenic, many affected individuals do not carry any one risk allele. Although in reality there will be a continuous spectrum between allele frequency and penetrance, most genetic findings have been at the two extremes; Table 1 lists some of the key differences between common risk variants and monogenic mutations. Two related striking features of discoveries from GWASs have emerged: first is the revelation of just how polygenic common diseases are; despite the identification of 70 variants associated with type 2 diabetes, 185 with lipid levels, 40 with heart disease and 180 with adult height, the vast majority of heritability remains unexplained. Second is how small the effect sizes are and how this has been reflected in the sample sizes required to detect associations, eg most of the 180 known variants associated with height required a GWAS sample size of 133,000 individuals and replication in an additional 50,000.2
Key differences between known common and rare disease variants.
Genome-wide association study findings in metabolic disease
These common variant associations currently have limited relevance to clinical decision-making. There are few examples where GWAS findings have proven useful to individual patients. The vast majority of variants identified by GWASs are common and of subtle effect. With the exception of the HLA alleles in autoimmune and inflammatory diseases, and some pharmacogenetic effects, the risks (usually expressed as odds ratios) associated with common alleles are <2.0, and for continuous traits such as body mass index (BMI) usually <0.1 standard deviation (SD), eg the most strongly associated variants associated with type 2 diabetes (the variant in TCF7L210) and coronary heart disease (the variant near CDKN2A/B11) confer risks, expressed as odds ratios of approximately 1.4 per risk allele. The variants most strongly associated with BMI and height do so with per allele effects of 0.1 SD (approximately 0.4 kg/m2) and 1 cm respectively.12 Most of the variants confer much smaller effects than these examples. Many studies have combined information from multiple associated SNPs and shown that these explain more of the phenotype.9,13,14 However, even when combined, the variants rarely provided sufficient statistical power to offer any predictive value, eg when considering the sensitivity and specificity of information from common genetic variants, the 40 strongest type 2 diabetes variants have a receiver operator curve (ROC) area under the curve (AUC) value of 0.63,13 where 0.5 is the same as flipping a coin and 0.8 is considered clinically useful. Age at menopause is another common trait where predictive value could be very useful for women planning families, because fertility starts to decline sharply 10 years before the menopause and women are tending to have their families later. However, the four common variants most strongly associated with age at menopause and, in one case, reducing the age at menopause by 1 year per allele still do not provide useful predictive power, with an ROC AUC of 0.60 for early menopause (<45 years).15
There are some common diseases where directly genotyping sets of common variants could be useful to individual patients. Multiple common variants may prove useful in identifying individuals with coeliac disease16 and different types of rheumatoid arthritis.17 Over the next few years, it will be worth noting how additional discoveries in the inflammatory and autoimmune diseases, where genetic effects tend to be stronger, help clinicians and their patients.
What are the advantages of genome-wide association studies?
If there is no direct benefit to individual patients, what have GWASs achieved? To further complicate this question, the common variants identified by GWASs do not necessarily represent the causal allele, and do not usually identify which gene is involved at any locus. Many tens of common variants are often strongly correlated due to linkage disequilibrium, and this means that we cannot usually say which one is causal out of the many that have been inherited together as part of the same ancestral piece of DNA (an example is given in Fig 1). Furthermore these clusters of associated common SNPs often occur in non-coding regions of the genome, deep in introns, outside genes or sometimes overlapping many genes. Dissecting which causal allele and which gene are the target of that causal allele has proven difficult. However, several lines of evidence strongly suggest that the causal gene is usually one of the two or three closest genes, eg the regions of the genome identified by a GWAS as associated with type 2 diabetes are enriched for monogenic diabetes genes such as HNF1A, HNF1B and PPARG, and small non-coding regions of the genome (enhancers) critical for islet-specific gene expression.18 Regions of the genome identified by GWASs as associated with height and altered lipid levels are enriched for monogenic genes, where mutations cause severe changes in height2 or lipid levels19 respectively (Table 2).
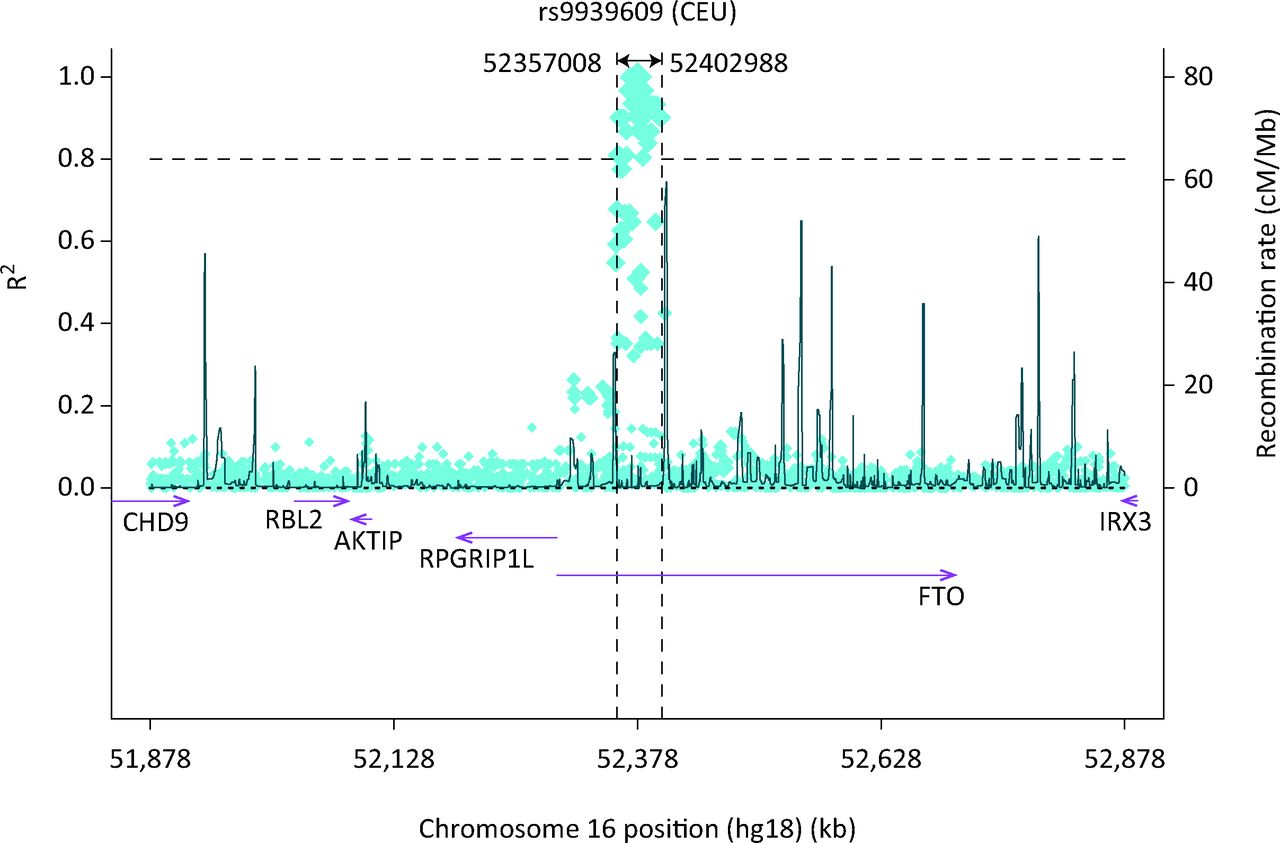
The region on chromosome 16 containing the FTO and IRX genes, the single nucleotide -polymorphism (SNP) most strongly associated with obesity and body mass index (BMI) (rs9939609) and the correlations with other SNPs in the region caused by linkage disequilibrium.
Allelic spectrum of known genes: examples of genes and loci where rare monogenic mutations and nearby common SNPs influence a very similar disease or trait.
Despite the limitations of GWASs, the robustness of the associations has provided an important first step in understanding biology – the genome-wide, unbiased nature of the genetic mapping efforts means that many new unexpected genes and loci have been highlighted. Here I outline several examples in metabolic disease.
The ‘fat gene’: FTO or IRX3?
In 2007 one of the first GWASs for type 2 diabetes discovered a common variant in an intron of the gene FTO which was associated with BMI and obesity.20–22 Carriers of two copies of the minor allele (16% of the population, the allele frequency being 40%) were on average 0.4 kg/m2 larger. Studies in children showed that this association was primarily driven by differences in adiposity.20 Since then hundreds of studies have tried to understand the role of the FTO protein in body weight regulation, with a combination of evidence from its expression in the brain23 to mouse transgenic studies,24,25 providing perhaps the most robust evidence that the obesity risk allele may operate through a gain-of-function mechanism on FTO. However, very recently, a new study, using a combination of human and mouse data, provides a case that the next-door gene, IRX3, is also involved in body weight regulation and may be the target gene of the obesity-associated SNPs.26 Further studies are needed to establish which, or whether both, of FTO and IRX3 are the causal genes. Either way, molecular studies of both genes have revealed more biological evidence for their role in body weight regulation, where previously there was none.
Genetic evidence for a metabolically obese–normal weight phenotype
Findings from GWASs have provided insight into the role of adiposity and metabolic traits in disease. These findings include that of a variant near the IRS1 gene. Although it is not known if the variant operates through IRS1, it is a strong candidate given the role of IRS1 in insulin signalling downstream to the insulin receptor. The variant was discovered through a GWAS of body fat percentage, and intriguingly the allele that predisposed to increased body fat percentage was associated with improved metabolic health, in the form of reduced circulating triglycerides, raised high-density lipoprotein (HDL), raised adiponectin levels27 and reduced risk of type 2 diabetes.28 The pattern of associations observed with the IRS1 allele provides genetic evidence for the so-called ‘metabolically obese–normal weight’ phenotype (sometimes turned around and referred to as the ‘metabolically normal–obese phenotype’).29 Researchers have hypothesised that individuals less able to store fat subcutaneously, due to reduced adipocyte differentiation or plasticity, may be thinner, but metabolically less healthy than others because they will store more fat viscerally – in the liver in particular.
Common variants identified by genome-wide association studies may modify monogenic phenotypes
Several studies have examined the effects of carrying multiple common, subtle effect alleles on monogenic disease traits. The penetrance of monogenic mutations in BRCA1 and HNF1A30 is increased in the presence of multiple common alleles predisposing to breast cancer and type 2 diabetes, respectively, eg women with a BRCA1 mutation and carrying more common breast cancer risk alleles than average (based on alleles from seven SNPs) are at greater risk of developing breast cancer than BRCA1 carriers with relatively few common risk alleles.31,32 More studies may result in clinical use of this information.
Summary
Before 2007 the number of common genetic variants reproducibly associated with common diseases and traits was fewer than 20. There are now many hundreds of variants reliably associated with all types of diseases and traits, from male pattern baldness to height to common disease predisposition, including metabolic disease, autoimmune disease and germline predisposition to cancer.
Despite this success at identifying variants, the GWAS findings are not generally clinically useful to individual patients. Instead they represent a first step towards improved understanding of disease aetiology.
Key points
Disease and trait altering variants discovered by genome-wide association studies (GWAS) tend to be common and of low penetrance
Loci identified by GWAS are enriched for monogenic genes relevant to the disease or trait
GWAS identifies variants, not genes, but the causal genes are likely to be very close in most regions
Unlike candidate gene and linkage-based studies GWAS approaches have provided very robust, reproducible findings
For most diseases, we likely need to discover more variants before GWAS findings can be translated into benefits to individual patients
- © 2014 Royal College of Physicians
References








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.