
ABSTRACT
Primary immunodeficiencies (PID) are a group of rare inherited disorders that manifest as heightened susceptibility to infection, autoimmunity and/or malignancy. By exploring their genetic and cellular aetiology, we can learn much about the basis of pathogen-specific immunity in humans. This is exemplified by mycobacterial susceptibility, which occurs across several types of PID, either as an isolated problem or as part of a broader pattern of susceptibility to infection. These experiments of nature have contributed to our understanding of the central role of T cells in activating infected macrophages to eliminate phagosomal mycobacteria through mutually activating, cytokine-dependent interactions. In recent years, the discovery of novel forms of PID has emphasised the important role of dendritic cells and monocytes in mycobacterial defence in humans. Here, we provide a brief overview of these new disorders alongside other genetic causes of susceptibility to mycobacterial disease.
Mycobacterial disease
Mycobacterium spp. are environmentally ubiquitous bacilli responsible for a significant burden of global disease. A spectrum of virulence runs across the genus: Mycobacterium tuberculosis (MTB) kills approximately 1.3 million people per annum, whereas Mycobacterium leprae continues to cause disfiguring disease. The less pathogenic non-tuberculous Mycobacteria (NTM) are mainly opportunistic pathogens of patients with structural lung disease or immunodeficiency; similarly, the live-attenuated vaccine strain of Mycobacterium bovis Bacille Calmette–GuÈrin (BCG) rarely causes disseminated disease (termed ‘BCG-osis’ 1) in the absence of immunodeficiency.
Mycobacterial pathogenesis
Insights into mycobacterial pathogenesis have come mostly from animal models and patient studies and, although we have a broad concept of pathogenic mechanisms, significant gaps remain (reviewed in Philips and Ernst2). Macrophages are the archetypal target cell for Mycobacterium spp., although other phagocytes (including neutrophils, monocytes and dendritic cells (DC)) also ingest bacilli2 and can support replication.3 Uptake is conditional for mycobacterial virulence and mycobacteria take advantage of considerable redundancy in the process of phagocytosis to enter target cells. Mycobacteria survive and replicate within phagosomes, autophagosomes and possibly also the cytosol2 of macrophages. The classical T helper 1 (TH1) cytokines, interferon-γ (IFN-γ) and tumour necrosis factor-α (TNF-α), produced by interleukin (IL)-12-polarised CD4+ T cells are required to activate macrophages and enable mycobacterial killing within the phagosome.2 The role of antigen-presenting cells in this process lacks clear definition (see below), although MTB has multiple mechanisms for downregulating major histocompatibility complex (MHC) class II expression in infected cells,4 suggesting that evasion of antigen presentation to CD4+ T cells is advantageous.
Mycobacterial susceptibility
Around ten times more people are infected with MTB than develop clinical tuberculosis (active TB), proving that the immune system, although unable to eradicate infection, often contains it sufficiently to prevent disease. Mycobacterial susceptibility is a feature of a range of clinical immunodeficiency syndromes. For example, the annual risk of active TB in patients with acquired immunodeficiency syndrome (AIDS) because of human immunodeficiency virus (HIV-1) infection is the same as the lifetime risk in the immunocompetent.5 Heightened susceptibility to mycobacterial disease is also a feature of several types of primary immunodeficiency (PID) caused by single-gene defects in immune function. Reflecting the frequency with which different Mycobacterium spp. are encountered, these disorders typically present not with MTB but with disease caused by its less pathogenic relatives, NTM and BCG. Efforts to define the molecular basis of these disorders have taught us important lessons about human mycobacterial immunity, including the crucial role of T cells, dendritic cells and the IL-12/IL-23–IFN-γ–signal transducer and activator of transcription 1 (STAT1) circuit.
Aims and scope
In this review, we summarise data on monogenic disorders that confer enhanced susceptibility to mycobacterial disease, with a particular focus on the recently described dendritic cell deficiency syndromes, and reflect on the fundamental insights into the nature of protective mycobacterial immunity that these ‘experiments of nature’ provide. Population-based genetic studies of single nucleotide variants (also known as single nucleotide polymorphisms) linked to mycobacterial susceptibility and data from murine models are outside the scope of this article.
Single gene factors in mycobacterial disease susceptibility
The analysis of monogenic immune defects has been a rich source of information about non-redundant cell types and molecular pathways in the human immune response to pathogens,6 by turns validating7 or challenging8 findings in animal models. Gene defects have been elucidated by various means, including linkage analysis, candidate gene studies and unbiased approaches, such as whole-exome sequencing.
Mycobacterial disease occurs as part of a broader pattern of susceptibility to infection in several forms of PID. The central role of T cells is illustrated by the susceptibility of patients with severe combined immunodeficiency to disseminated BCG disease, among many other infections.9 Chronic granulomatous disease (CGD), a monogenic disorder of the oxidative burst resulting from several mutations in NADPH oxidase,10 produces vulnerability to a specific range of intracellular microorganisms (including Mycobacterium spp. in patients from endemic regions),11 reinforcing the importance of intraphagosomal killing in the effective control of mycobacterial infection.
Another class of PID to inform our understanding of mycobacterial immunity is the rare Mendelian susceptibility to mycobacterial disease (MSMD) (reviewed in Casanova and Abel12). In the western world, most of these patients present with BCG-osis or NTM disease (although also at increased risk of TB). In contrast to classic forms of PID, patients with MSMD are often otherwise healthy, despite some increased susceptibility to other intramacrophagic pathogens (including disseminated non-typhoidal salmonella). Candidate gene studies led to the discovery of several autosomal recessive and X-linked inherited gene variants in the classic TH1 circuit: IL-12 p40 subunit (IL12B), and IL-12 receptor beta-subunit (IL12RB1), interferon-γ receptor 1 (IFNGR1) and interferon-γ receptor 2 (IFNGR2) and STAT1. These lesions are believed to disrupt the mutually activating interaction between the macrophage and T cell that culminates in the engagement of intraphagosomal killing mechanisms.
Although often considered as a form of MSMD, PID associated with mutations of nuclear factor-kappa B-essential modulator (NEMO) or IKBA (NFKBIA) typically produces a broader infectious phenotype (reviewed by a number of authors12–15). Therefore, it is all the more striking that iatrogenic blockade of the same pathway with anti-TNF to treat autoimmune disease is a well-documented cause of TB reactivation. More recently, MSMD-associated variants have been identified in CYBB16 and interferon-stimulated gene 15 (ISG15).17 Although CYBB is the gene classically mutated in X-linked CGD, Bustamante et al identified MSMD kindreds with distinct allelic variants that led to a milder, macrophage-restricted defect in the oxidative burst.16 More intriguingly, although ISG15 is necessary for antiviral immunity in mice,18 the reported human phenotype displayed preserved antiviral immunity and a specific defect in the IFN-γ response to mycobacteria,17 reinforcing the concept that monogenic immune defects in humans and mice can have divergent effects. It is likely that many more relevant mycobacterial susceptibility variants exist and will be identified in the coming years because up to half of patients with MSMD do not have a molecular diagnosis.14
DC deficiency in mycobacterial susceptibility
Dendritic cells (DC) are specialised antigen-presenting cells of the mononuclear phagocyte system that have unique functional capabilities, including lymph node antigen trafficking and potent activation of naÔve T cells.19 In capturing, processing and presenting antigen to T cells, DC are the main conduit between the innate and adaptive immune systems and, therefore, their role in the human immune system has been viewed as central. Until recently, no DC deficiency syndromes had been discovered to test this assumption. Functional defects in antigen presentation have been described; for example, MHC class II deficiency (known as bare lymphocyte syndrome) or Wiskott–Aldrich syndrome (which causes a defect in immune synapse formation). In addition, numerical DC deficiency is a feature of PIDs characterised by broad cytopenias (eg reticular dysgenesis, Ikaros deficiency and C-X-C chemokine receptor type 4 (CXCR4) deficiency (WHIM syndrome)). However, none of these conditions inform the specific role of the DC. Below, we describe the molecular basis and characteristics of three recently discovered monogenic DC deficiencies that are associated with mycobacterial susceptibility. To put these findings into context, we first summarise human DC development.
DC development in humans
Understanding of the development and functional specialisation of human DC lags behind that of the mouse. Ethical constraints mean that most human studies are performed in peripheral blood or using in vitro monocyte-derived DC systems that might have limited relevance to tissue in vivo. There is also limited consistency of cell surface markers used to define subpopulations of DC across mouse and human. As such, it is difficult to infer from mouse the facts of human DC development.
With the probable exception of self-renewing skin-resident Langerhans cells and some central nervous system resident mononuclear phagocytes, human DC subsets (including various subsets of myeloid DC, and plasmacytoid DC; Table 1) are derived from haematopoietic progenitor cells (both granulocyte-macrophage progenitors and multilymphoid progenitors20) in the bone marrow.19 This conclusion is supported by data from haematopoietic stem cell transplantation (HSCT) in humans.21
Human dendritic cell subtypes.a
There is no evidence that monocytes differentiate into DC in the steady state in humans and it is currently unclear what relation blood DC subsets have to those in tissues19,22 (Table 1). Committed DC precursors have not been found in either blood or bone marrow and it appears that tissue DC are derived from blood DC and/or CD34+ progenitor cells in the blood.19 In this context, in natura human DC deficiency can also teach us about DC development.
DC deficiency
Historically, DC numbers were not measured in clinical diagnostic laboratories and this might account for the delayed recognition of DC deficiency syndromes in practice (associated monocytopenia is a hint). Novel algorithms aid DC profiling in peripheral blood23 and can lead to more efficient recognition of DC deficiency. There are currently three recognised genetic DC deficiency syndromes (Table 2), which are characterised by deficiency of DC subsets in the tissues and/or peripheral blood together with other haematological and clinical features.22
Dendritic cell deficiency syndromes.
DCML because of GATA2 mutation
The most frequently encountered DC deficiency syndrome to date is so-called DC, monocyte, B and NK lymphoid deficiency (Dendritic cell, monocyte, B and NK lymphoid (DCML) deficiency24) resulting from autosomal dominant mutations in GATA-binding factor 2 (GATA2).25,26 This broad mononuclear cell deficiency has also been labelled ‘MonoMAC syndrome’ because of its association of heightened susceptibility to NTM infection (specifically M. avium complex (MAC)) with autosomal dominant or sporadic monocytopenia.27 This is a progressive disorder and, by the time mycobacterial susceptibility is evident, blood and tissue DC are typically absent, although macrophages and Langerhans cells are preserved.28 Associated features might include susceptibility to viral and fungal infections, autoimmunity, lymphoedema, myelodysplasia that might transform to acute myeloid leukaemia and pulmonary alveolar proteinosis (a rare disorder of alveolar macrophage function leading to alveolar protein accumulation and respiratory failure).28,29
Candidate gene studies26 and exome sequencing25 conclusively established that DCML/MonoMAC is caused by heterozygous mutation of the transcriptional regulator GATA2. Haploinsufficiency of this gene impairs normal haematopoietic development, resulting in dramatic loss of multilymphoid progenitors and partial loss of granulocyte-macrophage progenitors in bone marrow.24 Despite this, most patients are healthy in their first one to two decades and do not experience problems with BCG. It has been proposed that preserved immune cell numbers (granulocytes, T lymphocytes, plasma cells and class-switched B cells together with tissue macrophages and dermal Langerhans cells) confer sufficient immunity to deal with most childhood infections.22 It perhaps also implies a redundant role for DC in antigen presentation30 and detailed analysis of the function and specificity of CD4+ T cells in patients with DCML will help to clarify the degree of antigen presentation defect(s) and their consequences. Many patients with GATA2 deficiency are susceptible to mycobacterial disease (particularly NTM) and their cells exhibit defective IL-12 and IFN-γ responses to toll-like receptor (TLR) agonists in vitro.24 However, the precise contribution of specific cell types (eg DC versus monocytes) is impossible to dissect.
DC deficiency resulting from IRF8 mutations
The discovery of two distinct DC deficiency syndromes resulting from mutations in IFN-regulatory factor 8 (IRF8)7 further clarifies the role of DC in mycobacterial immunity. The IFN regulatory factors are transcriptional regulators involved in haematopoiesis as well as in the response to IFN and TLR signalling leading to IL12B and nitric oxide synthase 2 (NOS2) transactivation;31 murine studies have shown Irf8 to be central to DC development.32 In humans, two mutations have been described in the IRF8 DNA-binding domain7 (Table 2): the AR mutation K108E caused an isolated severe deficiency of DC and monocytes with associated myeloproliferation and profound immunodeficiency requiring HSCT; the AD mutation T80A was instead associated with MSMD and an apparently specific deficiency of circulating CD11c+ CD1c+ myeloid DC.7 The latter syndrome seems to offer an important test of the role of DC in mycobacterial immunity, but poses several questions. Perplexingly, the ‘missing’ DC subset is not that typically associated with cross-presentation and neither does it correspond to the CD8α DC subset that is preferentially lost in murine Irf8 deficiency. Furthermore, BCG responsiveness was surprisingly preserved in patient peripheral blood ex vivo, although a defect in IL-12 production in response to TLR7/8 stimulation was observed in patients with AD T80A compared with controls. One interpretation is that mycobacterial susceptibility in AD T80A is the result of failure of IL-12 production specifically by this CD11c+ CD1c+ DC subset, similar to defects in IL12p40 or IL12R (see above). Perhaps in keeping with this, CD1c-restricted T cells with mycobacterial specificity have been reported33 and might require CD1c+ myeloid DC for their development and priming. The impact of AD IRF8 deficiency on mycobacterial killing within infected phagocytes merits further investigation as another potential mechanism of increased susceptibility.
Perspectives on DC development and function
The relative preservation of tissue macrophages and Langerhans cells in the face of the profound monocytopenia that accompanies deficiencies of GATA2 and AR IRF8 implies a distinct developmental origin for these cells.22 Although both disorders impair DC development in the bone marrow, the accompanying context differs dramatically: the K108E mutant results in accumulation of myeloid-lymphocyte precursors (MLP) and granulocyte-myeloid precursors (GMP),7 whereas GATA2 deficiency causes a marked deficit of MLP (and to a lesser degree GMP).24 This indicates that these molecules function at distinct points in DC differentiation, with GATA2 acting proximally and IRF8 distally in the MLP/GMP stage.
Interestingly, CD4+ T cells from the patient with AR IRF8 deficiency exhibited impaired production of various cytokines (including IFN-γ and IL-17) to CD3/CD28 stimulation ex vivo, suggesting a functional T cell defect in vivo. 7 In keeping with this interpretation, an unusually high proportion of naÔve cells in the T cell compartment of this patient was observed.7 Further studies to define the T cell phenotype in both DC deficiency syndromes will shed light on the possible effects of impaired antigen presentation on the T cell repertoire.
Summary
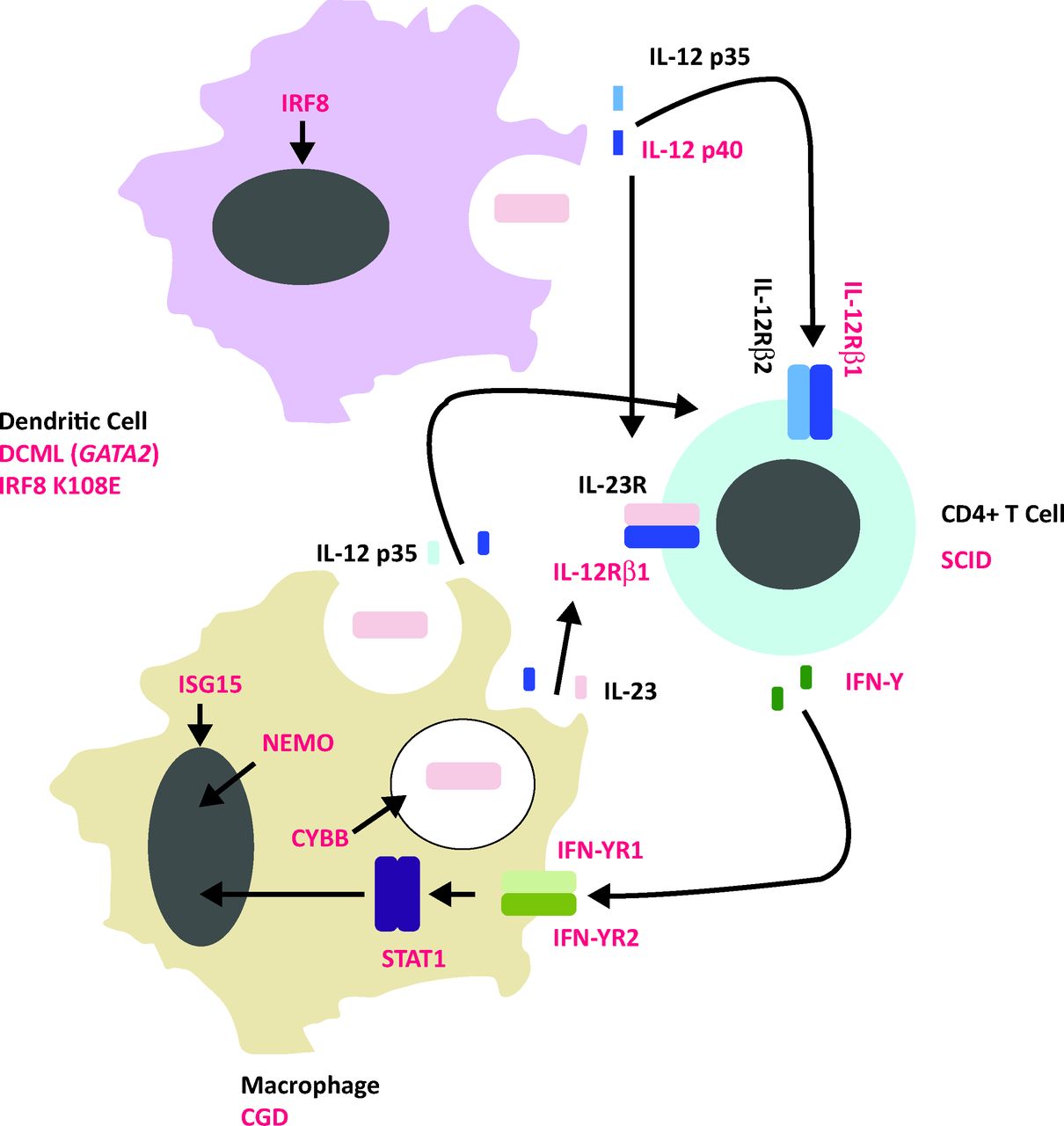
The identification of DC deficiency in the context of mycobacterial susceptibility adds a missing piece to the jigsaw of pathogen-specific immunity in humans (Fig 1). To the conventional diagrammatic representation of the mycobacterially infected macrophage communing with an antigen-specific T lymphocyte, we must add a third player: the DC. Future studies in these rare patients should seek to disentangle the respective contributions of DC, monocyte and T cell to functional immunodeficiency and could inform attempts to bolster adaptive immunity by smarter approaches to vaccination. ?

Model of mycobacterial susceptibility. Single gene variants in the molecular components in red type are implicated in mycobacterial susceptibility in humans. In addition, numerical or functional cellular deficiency syndromes linked to mycobacterial susceptibility are outlined below the relevant cell type. CGD = chronic granulomatous disease; DCML = dendritic cell, monocyte, B and NK lymphoid deficiency; IFN = interfeuron; IL = interleukin; IRF8 = interferon-regulatory factor 8; ISG15 = interferon–stimulated gene 15; NEMO = nuclear factor-kappa B-essential modulator; SCID = severe combined immunodeficiency -syndrome; STAT = signal transducer and activator of transcription 1.
- © 2014 Royal College of Physicians
References








{kind=link}
Jump to section
- Article
- ABSTRACT
- Mycobacterial disease
- Mycobacterial pathogenesis
- Mycobacterial susceptibility
- Aims and scope
- Single gene factors in mycobacterial disease susceptibility
- DC deficiency in mycobacterial susceptibility
- DC development in humans
- DC deficiency
- DCML because of GATA2 mutation
- DC deficiency resulting from IRF8 mutations
- Perspectives on DC development and function
- Summary
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.