
ABSTRACT
Our knowledge of the morphological heterogeneity of cancer has recently been augmented by the genomic heterogeneity revealed by the use of next-generation sequencing technology. We now know that no two cancers are alike and that even different regions within the same tumour vary in their composition. Tumours consist of multiple clonal populations and they evolve under Darwinian principles. This review summarizes some of the causes of such diversity and its implication for cancer management.
Cancer as a clonal disease
Over a century and a half has elapsed since Rudolf Virchow first benefited from the development of light microscopy and deduced the cellular origin of cancer. Gross pathological examination of cancerous masses was surpassed by histological description of the disordered cellular appearance and growth patterns characteristic of the malignant process. Following the discovery of proto-oncogenes in human DNA by Harold Varmus and colleagues, it was demonstrated that cancer was a disease of the DNA.1 These proto-oncogenes could become oncogenes following genetic mutations or overexpression, resulting in abnormal function of their respective protein products. Since many of these oncogenes are cell surface receptors or intracellular kinases, great hope was attached to the development of specific chemical inhibitors as a treatment for cancer.
Many targeted therapies are now in routine clinical use, including inhibitors of keys nodes in intracellular signalling networks, such as epidermal growth factor receptor (EGFR), mitogen-activated kinases such as BRAF, and the serine/threonine protein kinase mammalian target of rapamycin. However, the response to these therapies is not uniform, even if their use is restricted to patients with activating mutations in the relevant gene that should render them susceptible to such treatments. Differential responses between sites of metastasis within a single patient are common and periods of initial response are invariably followed by progression with a drug resistant clone. Peter Nowell was one of the first to suggest that cancer developed from a single mutant cell and that the accumulation of further somatic mutations in DNA resulted in disease progression akin to Charles Darwin's theory of gradual evolutionary change resulting in speciation.2
The development of next-generation sequencing (NGS) technologies and collaborations such as the 1000 Genomes Project, The Cancer Genome Atlas and the International Cancer Genome Consortium have enabled us to analyse the somatic mutations, copy number variations and structural rearrangements found in cancer at an unprecedented resolution. This has shown substantial diversity across and within cancer histotypes. For example, cancers such as acute myeloid leukaemia have a very low somatic mutation frequency, in comparison to more complex cancers such as squamous cell carcinoma of the lung and melanoma.3 However, tracing the evolutionary lineage of cancer clones back to an early cancer progenitor cell and the identification of genomic instability as an early driver of progression, resulting in an increased somatic mutation rate and errors in chromosome segregation during mitosis, was identified long before the development of NGS.4 The high throughput and resolution of NGS with improvements in computing power and data storage have been important developments in the last decade. This has enabled researchers to catalogue cancer genomes, create evolutionary trees to demonstrate how a cancer has developed over time, examine the temporality of different mutational processes and describe the ensuing heterogeneity that clonal evolution causes within a tumour.
Cancer evolution
The kinetics of tumour evolution is of great interest and holds significant clinical relevance. If cancers evolve in a linear fashion, when a cancer cell acquires an advantageous mutation this clone would theoretically sweep through the tumour, replacing previous clones. This would imply that after each clonal sweep, mutations in cancer-associated genes would be ubiquitously present throughout the tumour and a single biopsy would be representative of the somatic mutational landscape of the tumour. In practice, in the setting of advanced disease, a single biopsy of the most easily accessible lesion is used as reflective of the tumour burden as a whole. Current evidence suggests that the majority of cancers evolve in a branched or punctuated pattern that results in significant temporal and spatial heterogeneity. In adenocarcinoma of the pancreas, genetic instability caused by telomere dysfunction occurs early and drives tumour progression with further diverging patterns of clonal evolution in different metastasis, resulting in heterogeneity between the primary lesion and metastatic sites. In the patients studied all have some shared genomic rearrangements between their primary and metastatic lesions, but different metastatic sites develop private rearrangements causing loss of cycle control and activation of key oncogenes.5
Phylogenetic trees like those first conceived by Darwin can be used to represent tumour evolution and infer relationships between different cancer cell populations (Fig 1). Single cell sequencing in breast cancer and acute lymphoblastic leukaemia (ALL) has been used to demonstrate the extent of intratumour heterogeneity and draw evolutionary trees for these tumours. These groups used analysis of copy number changes to draw phylogenetic trees which show the relationship and evolution of the different subclonal populations within the tumour. In both scenarios they demonstrated that the tumours evolved by punctuated evolution with clonal expansion and branching phylogenetic trees rather than in a linear fashion.6,7

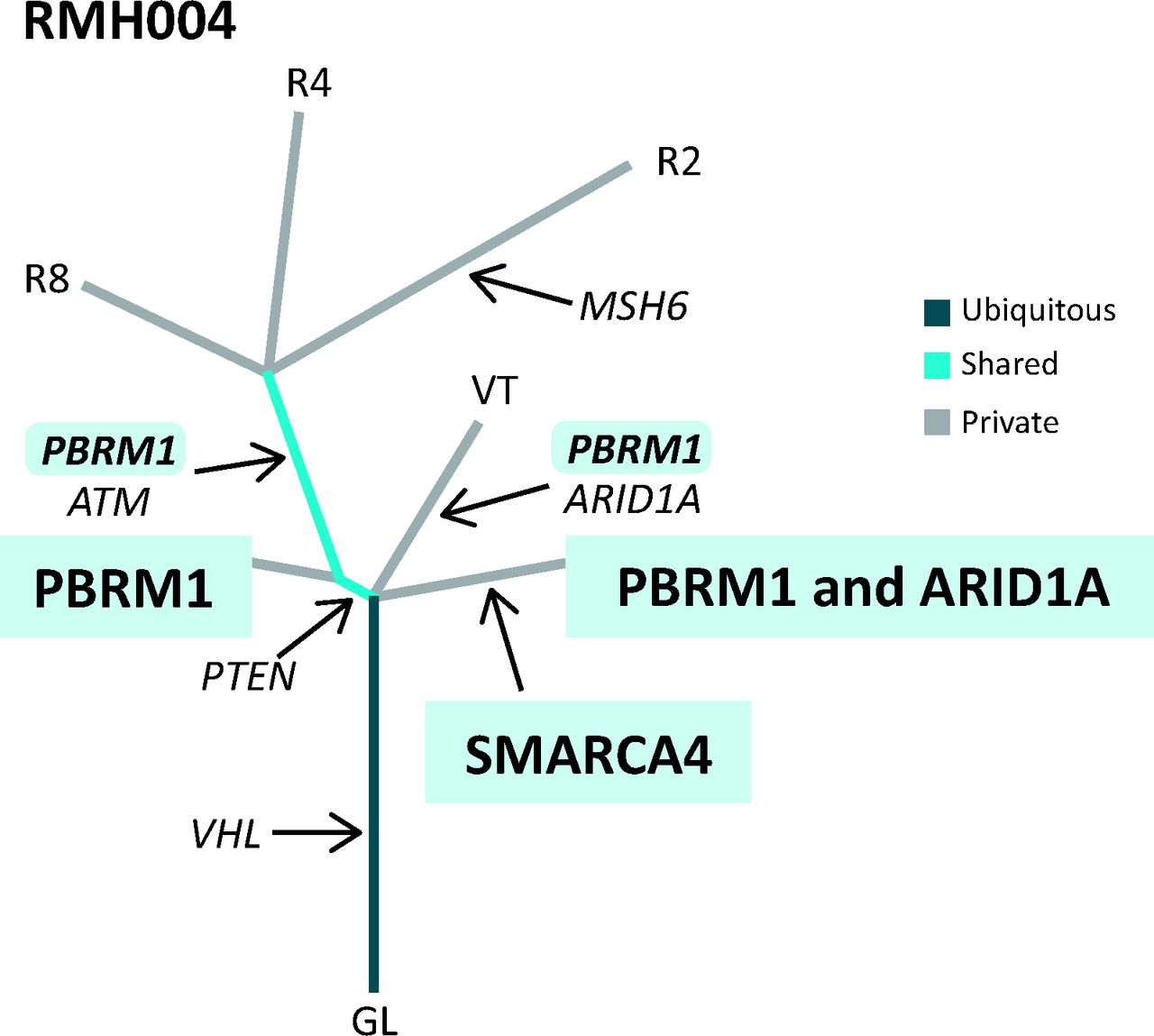
An example of a phylogenetic tree drawn from multi-region whole exome sequencing data from a patient with ccRCC with a clonal (trunk) mutation in VHL and subclonal (branch) mutations in PBRM1 and ARID1A. Distance of each branch represents evolutionary time and how different the tumour regions are from each other. ccRCC = clear cell renal cell carcinoma; GL = germline.
Causes of cancer evolution
In a study assessing the different mutational signatures found in cancer (patterns of mutations detected by NGS in large numbers of different tumour types), a wide number of factors such as smoking, age, UV radiation and defects in DNA repair genes generate diversity within tumours which contribute to increased genomic instability.8 Genomic instability is an enabling hallmark of cancer and a common footprint found in cancer genomes.9 This instability can occur through a diverse range of mechanisms leading to an elevated mutation rate, gene copy number changes or chromosomal instability resulting in aneuploidy.10
Defects in the genes MSH2 and MLH1 which repair mismatched DNA bases during DNA replication, as well as small insertions or deletions at areas of repetitive DNA sequences (microsatellites), are common in colorectal cancer and result in hypermutation and microsatellite instability.11 In clear cell renal cell carcinoma (ccRCC) mutations found in histone modifying genes, such as SETD2 and chromatin remodelling genes such as PBRM1, have been shown to be cancer drivers.12,13 These genes are involved in the process of gene transcription from tightly condensed genomic DNA and mutations in them may contribute to genomic instability. Evidence of whole genome doubling, a spontaneous event for which the aetiology remains unclear, can be found in many cancers. The process of genome doubling enables cancer cells to withstand errors in the segregation of chromosomes during mitosis and is associated with a worse prognosis in colorectal cancer.14 Surviving this event allows aneuploidy and chromosomal rearrangements that create clonal diversity within tumours. Chromosomal instability has been shown to be an indicator of a poor prognosis, presumably because the greater genetic diversity it affords allows tumours to better withstand the selection pressure of the tumour microenvironment and cancer treatments.15,16
Novel mechanisms for the evolution of cancer genomes have also recently been proposed. In prostate cancer, highly interdependent genome-wide translocations and deletions – termed chromoplexy – accumulate during oncogenesis and progression. These chromoplexy events occur relatively infrequently in the lifetime of the cancer, in a punctuated pattern, but deregulate multiple prostate cancer associated genes simultaneously.17 Shattering of whole chromosomes with disorganised rejoining of fragments – termed chromothripsis – has been shown to be a cause of several genomic lesions that can drive cancer formation. Although this is a relatively infrequent phenomenon, evidence of this process has been found in the majority of cancer types.18
Some studies have shown that genomic instability seen in advanced cancer may be iatrogenic. In a study of ALL, treatment was shown to be a transient inducer of genome instability with an increase in the mutational burden found in clones following treatment.19 Similarly in patients with glioblastoma multiforme (GBM), treatment with temozolomide, the current standard first line chemotherapy, was found to select for clones with defects in mismatch repair genes and accelerate the mutation rate following treatment.20 There are many contributors to tumour evolution, both cell-intrinsic (eg genomic instability) and exogenous (eg tumour microenvironment and cytotoxic chemotherapy), but because of the branched pattern of tumour evolution, significant heterogeneity is found within a single cancer which has implications for clinical practice.
Clinical implications of tumour evolution and tumour heterogeneity
Clonal evolution results in significant regional heterogeneity within individual tumours and their metastasis. Sequencing of the exomes of multiple regions from a ccRCC primary tumour and corresponding metastases shows significant allopatric separation of subclones (Fig 2). Loss of the long arm of chromosome 3 and mutations in von Hippel-Lindau (VHL) gene are common genetic aberrations seen in the ‘trunk’ of the branched evolutionary trees. Loss of VHL results in stabilisation and activation of hypoxia inducible factor 1 alpha, a transcription factor that induces a number of genes associated with cancer, whereas a number of mutations in other cancer associated genes, such as SETD2 and PTEN, are seen only in spatially restricted regions of the tumours and located on the distal branches of the phylogenetic tree.21,22 In these studies only two-thirds of the cancer-associated somatic mutation were shared between different regions in many tumours. The implications of these data are that using a single biopsy to look for biomarkers that guide clinical decisions is likely to result in significant sampling bias and may miss subclonal spatially separated cancer drivers in ccRCC. In this study over five biopsies would have been required to detect over 90% of the driver genes found in many of the tumours.
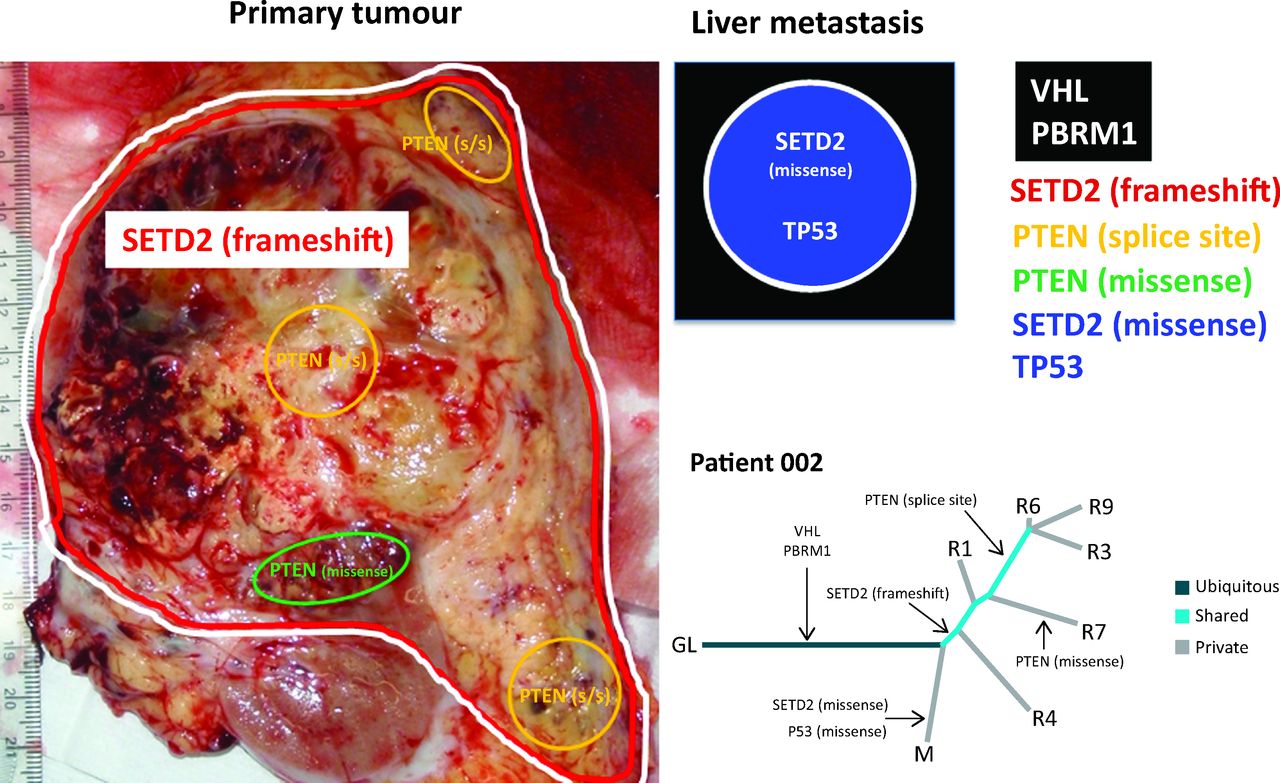
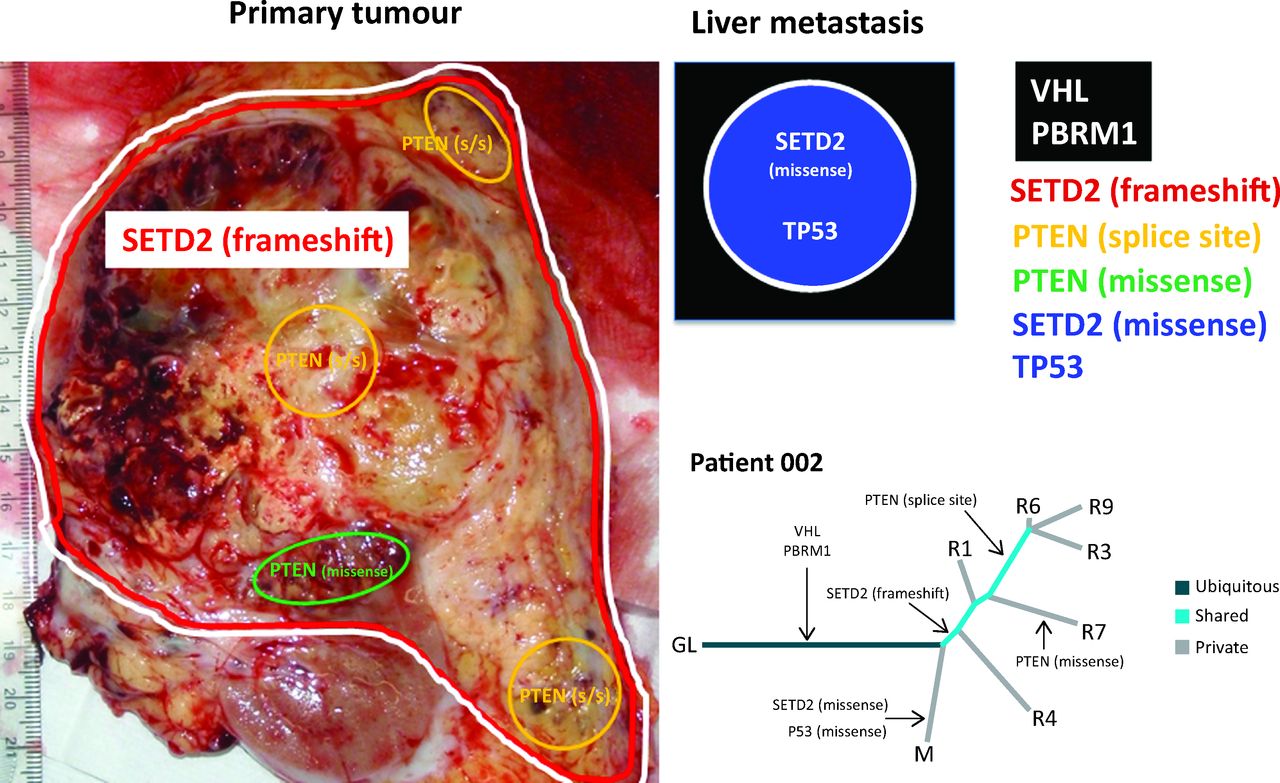
A representation of allopatric separation of cancer driver mutations determined from multiregion sequencing in a patient with ccRCC. In the primary tumour specimen, VHL/PBRM1 and SETD2 mutations are found throughout as indicated by the red and white lines. Within the primary tumour, two subclonal populations show convergence on PTEN with a splice site (s/s) mutation and a missense mutations indicated by yellow and green areas respectively. Although the PTEN splice site mutation is found in a greater number of regions in comparison to the missense mutation. The liver metastasis represented graphically in the black box has ubiquitous VHL and PBRM1 mutations that it shares with the primary tumour. However, this liver metastasis has developed a private missense mutation in SETD2, differing from the SETD2 frameshift mutation seen in the primary tumour, and a TP53 mutation. In the phylogenetic tree, R1, 3, 4, 6, 7 and 9 are different tumour regions and M is the liver metastasis. Evidence of convergent evolution is shown with a SETD2 frameshift and PTEN splice site mutation in the primary tumour. By contrast, a SETD2 missense mutation is seen in the liver metastasis and PTEN missense mutation seen in another tumour region. Tumour evolution occurs differently in the different regions of the primary tumour and liver metastasis. ccRCC = clear cell renal cell carcinoma; GL = germline; VHL = von Hippel-Lindau.
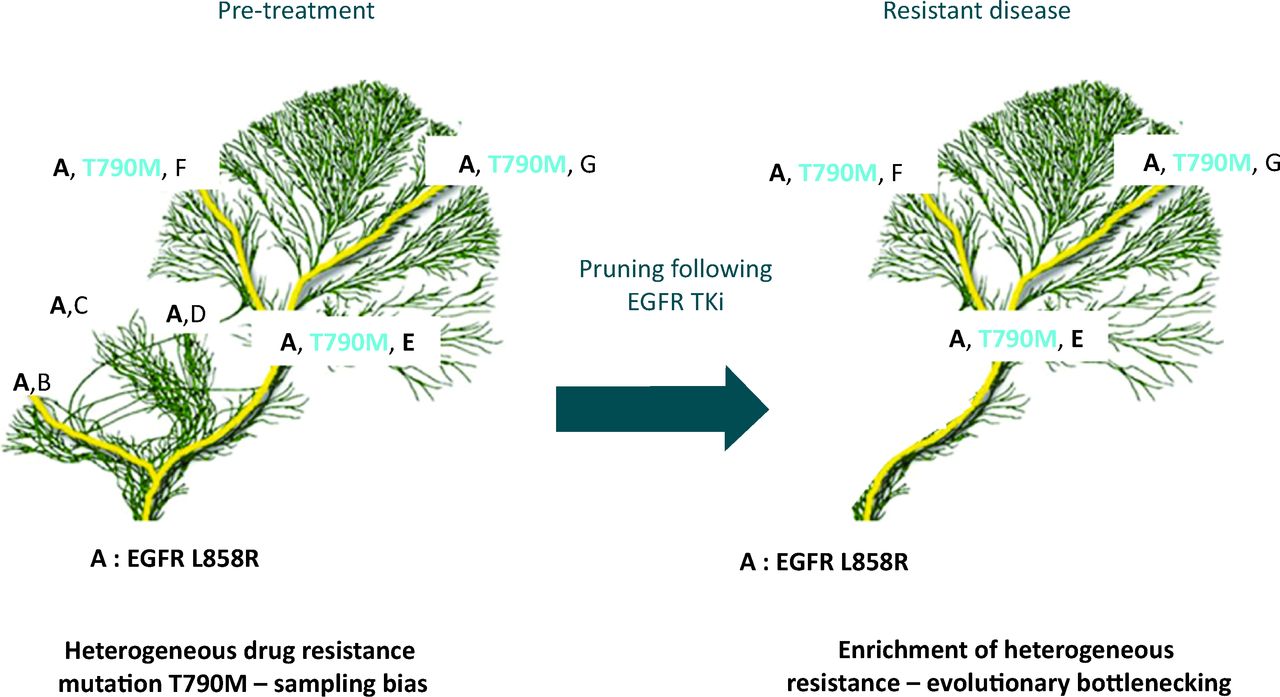
Temporal heterogeneity due to clonal evolution means that analysis of the primary may not be representative of metastatic lesions that have undergone different mutational processes and selective pressures.7,23 Basing treatment decisions on analysis of archival material from a primary tumour, as is common practice, may therefore not always be entirely representative of the disease at metastatic relapse. In a retrospective study of HER2 discordance between primary and metastatic lesions, over a quarter of patients with HER2-positive breast cancer lost expression of this biomarker in their metastatic relapse samples. If patients with discordant biomarkers had been given trastuzumab, which targets the HER2 protein, they had poorer overall survival.24 In NSCLC the EGFR T790M mutation is associated with resistance to standard first line therapy with EGFR tyrosine kinase inhibition (TKi). In a study using NGS, which has a greater sensitivity than current clinical diagnostic methods, temporal heterogeneity in response to treatment was seen following treatment of patients with EGFR TKi. The prevalence of clones containing the T790M resistance mutation increased following treatment as sensitive clones die on treatment with EGFR TKi, allowing clonal expansion of the resistant population.25 This results in pruning of the evolutionary tree with a general restriction of diversity, but selection of therapy resistant clone, akin to evolutionary bottlenecking (Fig 3). If bottlenecking in response to treatment can be reliably predicted in patients, or empirically tested, this transient lack of diversity may allow precision medicine with successful sequential treatments.
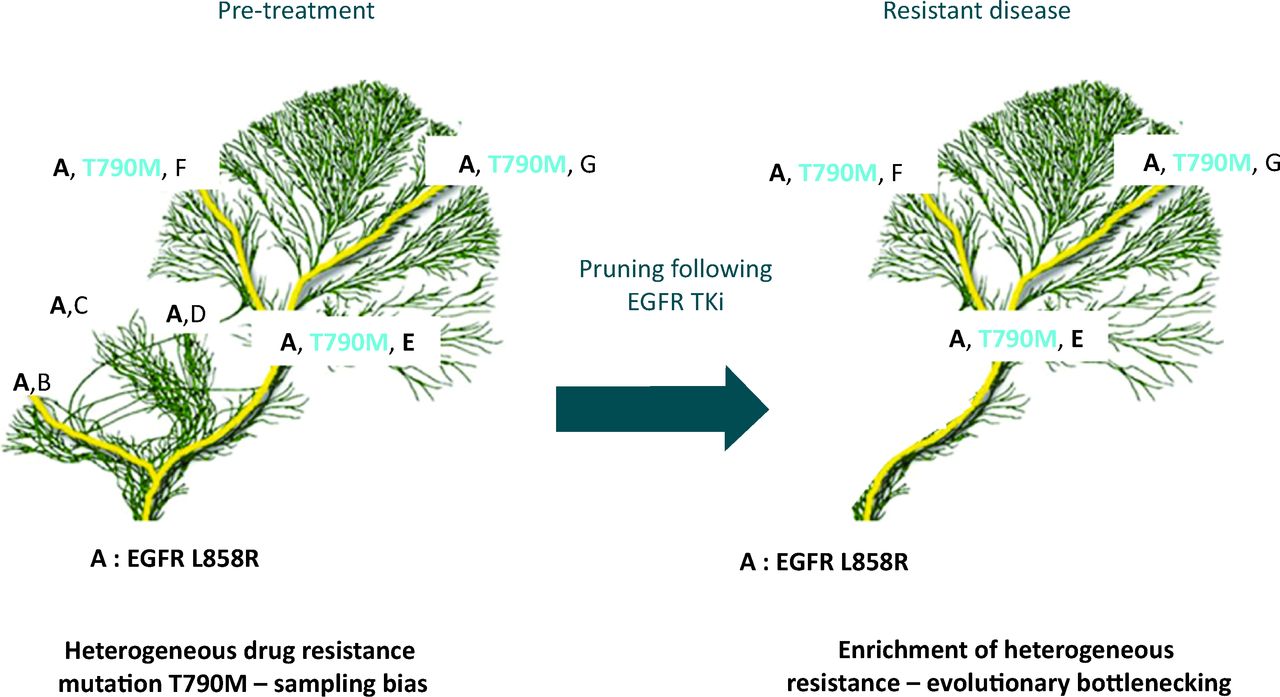
Tree and branch representation of tumour evolution in response to therapy. Ubiquitous mutation EGFR L858R in the tumour (mutation A) found in every subclone and every tumour region, represented in the trunk and branches of the tree. Diverse heterogeneous somatic events are represented by the branches and the leaves (mutations B, C, D, T790M, E, F and G). Multiple branches all contain the L858R EGFR mutation sensitising to EGFR TKi therapy but some branches contain the T790M resistance mutation. The presence of the resistance mutation would be missed by a single biopsy if geographically separated. Following treatment, drug sensitive branches are pruned from the tree restricting diversity, causing an evolutionary bottleneck with loss of mutations B, C and D, as they were found only in clones without the T790M mutation. Persistence of somatic events A, E, F and G are seen as they co-exist with the T790M mutation, conferring resistance to EGFR TKi. EGFR = epidermal growth factor receptor; TKi = tyrosine kinase inhibition.
In many current clinical trials in cancer the use of a targeted therapy is decided based on the presence of an actionable mutation in a gene that should render the patient's tumour sensitive to the drug in question. Because of spatial and temporal heterogeneity caused by clonal evolution, these actionable mutations may be present only in the branches of the phylogenetic tree and, although detected by chance on a single biopsy, not ubiquitous throughout the tumour. The presence of a subclonal driver gene has been shown to have a significant impact on outcome.21,25,26 An actionable mutation thought to be ubiquitous on a single biopsy may be present in only a small subclonal fraction; alternatively a driver gene associated with drug resistance may be present in a minor subclone which expands in response to treatment. Both scenarios would result in only a transient response to treatment, as is commonly seen in clinical practice. The clinical relevance of these subclonal drivers in NSCLC is being assessed in a multi-centre UK-based longitudinal observational study using multi-region sequencing of primary tumours at radical resection. The TRAcking Non-small Cell Lung Cancer Evolution Through Therapy (Rx) (TRACERx) trial will also collect cell free tumour DNA (cfDNA) and circulating tumours cells (CTCs) that are commonly found in the plasma of cancer patients, particularly those with a heavy burden of disease. Circulating ‘liquid biopsies’ are an attractive method for resolving such diversity and sampling bias witnessed in single biopsy studies of tumour biology. Deep sequencing of multi-regions from the primary tumour, cfDNA/CTCs and repeat biopsy samples at relapse and from some patients at autopsy will allow us to track tumour evolution. With this unprecedented level of knowledge regarding evolution in NSCLC, we aim to determine the clinical relevance of subclonal cancer drivers and define the origins of the lethal subclone that some patients will eventually succumb to in order to define strategies for timely intervention with systemic therapies that may improve outcomes.
Numerous gene-expression-based classification signatures have been published for cancer that are either prognostic for outcome or predict for response to particular treatments. Many reports subclassify a tumour type into subtypes based on these gene expression signatures. In GBM a gene expression signature has sub classified it into four categories: proneural, neural, classical and mesenchymal, which all respond differently to standard therapy.27 However multi-region gene expression analysis has shown that significant intratumour heterogeneity exists and that multiple regions of the same tumour can cluster into two or three of the different GBM subtypes.28 Tumour heterogeneity can dilute the predictive ability of these signatures when multiple drug resistance mechanisms are present in a single tumour, putting patients simultaneously into both good and bad prognostic groups depending on which region of the tumour is used for classification.21,29 These findings limit the utility of these classifiers which many had hoped could be used as stratification tools in future clinical trials to determine groups who are more likely to respond to specific therapies.
Conclusion
Clinical trials of targeted therapies in advanced metastatic disease have yielded disappointing results in the majority of cases. The knowledge that Darwinian evolutionary principles are in part responsible for tumour heterogeneity offers some explanation for the limited success of such approaches. Evolution is a consequence of both cell intrinsic phenomena, such as genomic instability, and extrinsic phenomena mediated by the tumour microenvironment and iatrogenic selection pressures.30 Future clinical studies will need to decipher the mechanisms responsible for cancer diversity and to improve cancer selection pressures either by targeting the clonally dominant drivers of cancer or resolving the importance of subclonal drivers to develop intelligent combination therapies.
- © 2014 Royal College of Physicians








{kind=link}
{kind=link}
{kind=link}