
ABSTRACT
Cancer is the ultimate disorder of the genome, characterised not by just one or two mutations, but by hundreds to thousands of acquired mutations that have been accrued through the development of a tumour. Thanks to the recent increase in the speed of sequencing offered by modern sequencing technologies, we are no longer restricted to exploring tiny fragments of protein-coding portions of the human genome. We can now read all the genetic material in human cells. Here, the framework of a next-generation sequencing experiment is explained, giving insight into the advances and difficulties posed by processing the enormous datasets generated through these methods. Some of the recent insights into tumour biology, that exploit the extraordinary surge in scale and the digital nature of next-generation sequencing, are highlighted, including cancer gene discovery, the detection of mutation signatures and cancer evolution. Technological and intellectual developments are starting to shape the personalized cancer genomic profiles of tomorrow. Let's train the next-generation of clinicians to be able to read them from today.
- Cancer genomics
- next-generation sequencing
- cancer genes
- mutation signatures
- cancer evolution
- tumour heterogeneity
Introduction
The genetic material in cells is prone to mutation. From the moment of conception, a fertilised egg containing a single copy of the human genome will undergo many thousand cell divisions, potentially acquiring mutations with each round of replication. In addition, the baby that is born and grows to adulthood will, through its life, be exposed to several endogenous DNA mutagenic processes, such as reactive by-products of cellular metabolism and enzymatic degradation, as well as a variety of exogenous DNA mutagens, such as ultraviolet radiation and various chemical compounds.
Human cancers are known to be highly mutated entities1 with marked genetic differences when compared with the original genome at conception. The genome of a cancer will carry all the mutations that have been acquired by the cell that became the malignant clone, which includes premalignant mutations as well as mutations accrued during tumourigenesis.1–3 Therefore, a cancer genome is a historical record of the mutagenic processes that have occurred through the life of the patient with cancer. A few of those mutations (<10) are thought to confer a selective proliferative advantage to the cell that first gave rise to the cancer clone and are referred to as ‘driver’ mutations.1 Most mutations in a cancer are simply bystander events, ‘passenger’ mutations that arise because of the damage that the cell has been subjected to through tumour development or because of the failure of cellular repair pathways to manage physiological quantities of damage.1–3 Despite not being causative for cancer, passenger mutations report on the biological perturbations that have occurred through the life of the patient with cancer.2,3
Using modern sequencing approaches to read cancer genomes
The advent of modern sequencing approaches, namely next-generation sequencing (NGS) technology,4 has resulted in an extraordinary increase in the speed and scale of sequencing the human genome. No longer are we restricted to exploring small polymerase chain reaction (PCR)-defined portions of the genome (<750 base pairs (bp) per PCR);5,6 current large-scale resequencing approaches are able to explore a subset of genes of interest (targeted sequencing), all protein-coding exons (exome sequencing) or even whole cancer genomes (whole-genome sequencing (WGS)), in a single experiment.7,8
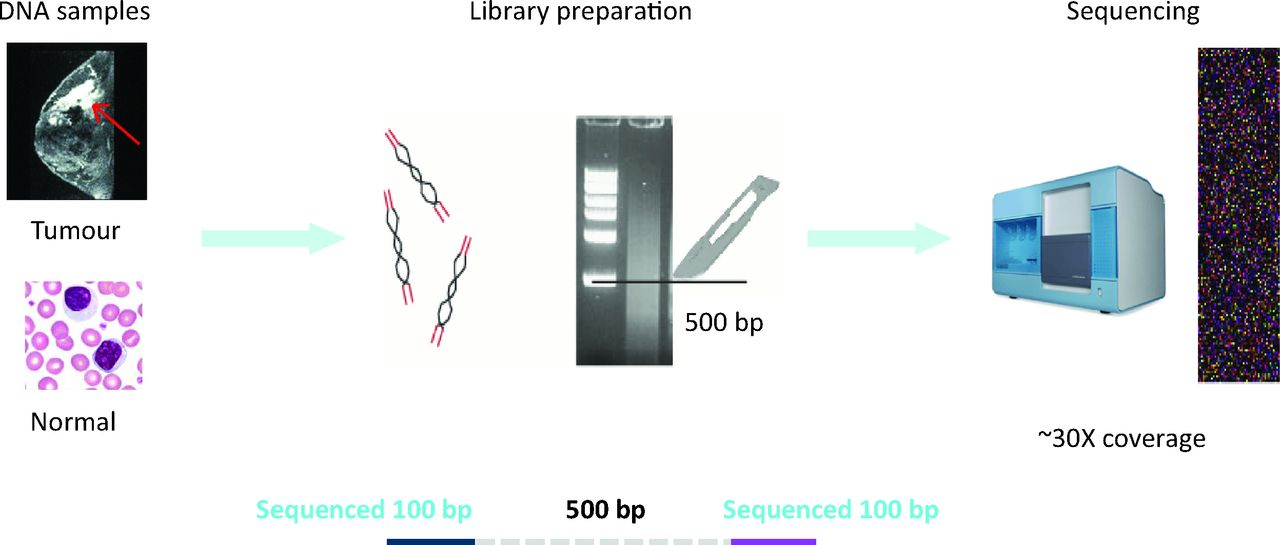
In essence, two samples are required per patient with cancer (Fig 1): a DNA sample from the cancer (ie ‘tumour’ DNA that is representative of the cancer clone, although some degree of heterogeneity within the cancer population is likely) and a DNA sample (ideally) extracted from peripheral blood lymphocytes (ie ‘normal’ DNA derived from a heterogeneous population of cells and representative of the germline genome). Each of the two DNA samples is subjected to independent fragmentation to generate many billions of DNA fragments per sample.4 A size-selection step is carried out to define the fragment size of interest (eg approximately 500 bp for WGS). Usually, 100 bp at each end of the 500-bp fragment will be sequenced using next-generation approaches to have read each base pair of the 3,000,000,000 bases present in the human genome, at least 30 times over, in each sample. This paired-end high-coverage NGS strategy is a general principle that can be modified (eg single-ended sequencing, 50, 75 or 150-bp read lengths or variable fragment sizes) depending on the nature of the experiment.

Principle of a paired-end NGS experiment. DNA samples are obtained from the cancer and from matched peripheral blood lymphocytes for each patient. In the library preparation phase, the DNA samples are independently fragmented into billions of pieces and prepared for the sequencing process (repair of ends of DNA fragments and ligation of sequencing adaptors). A size-selection step (which can now be performed using different methods and not just slicing following gel electrophoresis) is performed to obtain desired fragment sizes (here, 500 bp) to make a NGS library. Each library contains billions of fragments of DNA and is representative of the entire genome of the population of cells in each cancer-matched normal sample. In this method, 100 bp at both ends of each approximately 500-bp fragment is sequenced. Each library is sequenced to generate enough raw sequence to ensure an average -coverage of 30-fold per reference base in the genome. NGS = next-generation sequencing; bp = base pairs.
The raw outcome of a sequencing experiment is a dauntingly enormous collection of fragments of DNA that require alignment back to the reference genome to make a sensible and contiguous human genome (Fig 2).9 The basic tenet of identifying somatic mutations in cancer genomes relies on identifying differences in the genetic material of the tumour and normal sample relative to the reference genome (Fig 2). The tumour sample will contain acquired somatic mutations as well as germline variation (Fig 2). Subtraction of the germline variation identified in the normal sample will result in a final catalogue of somatic variants present in the cancer of a patient (Fig 2).

Principle of calling somatic mutations. (a) Millions of short NGS reads generated by sequencers are (b) aligned back to the -reference genome separately in tumour and normal genomes. (c) All differences detected when comparing the tumour with the reference genome include somatic (red crosses) and germline variants (purple crosses). (d) All differences in the normal genome relative to the -reference genome are identified independently (purple crosses). (e) The germline polymorphisms in the normal genome are subtracted from the tumour genome to generate the catalogue of somatic variants for each cancer of each patient. NGS = next-generation sequencing.
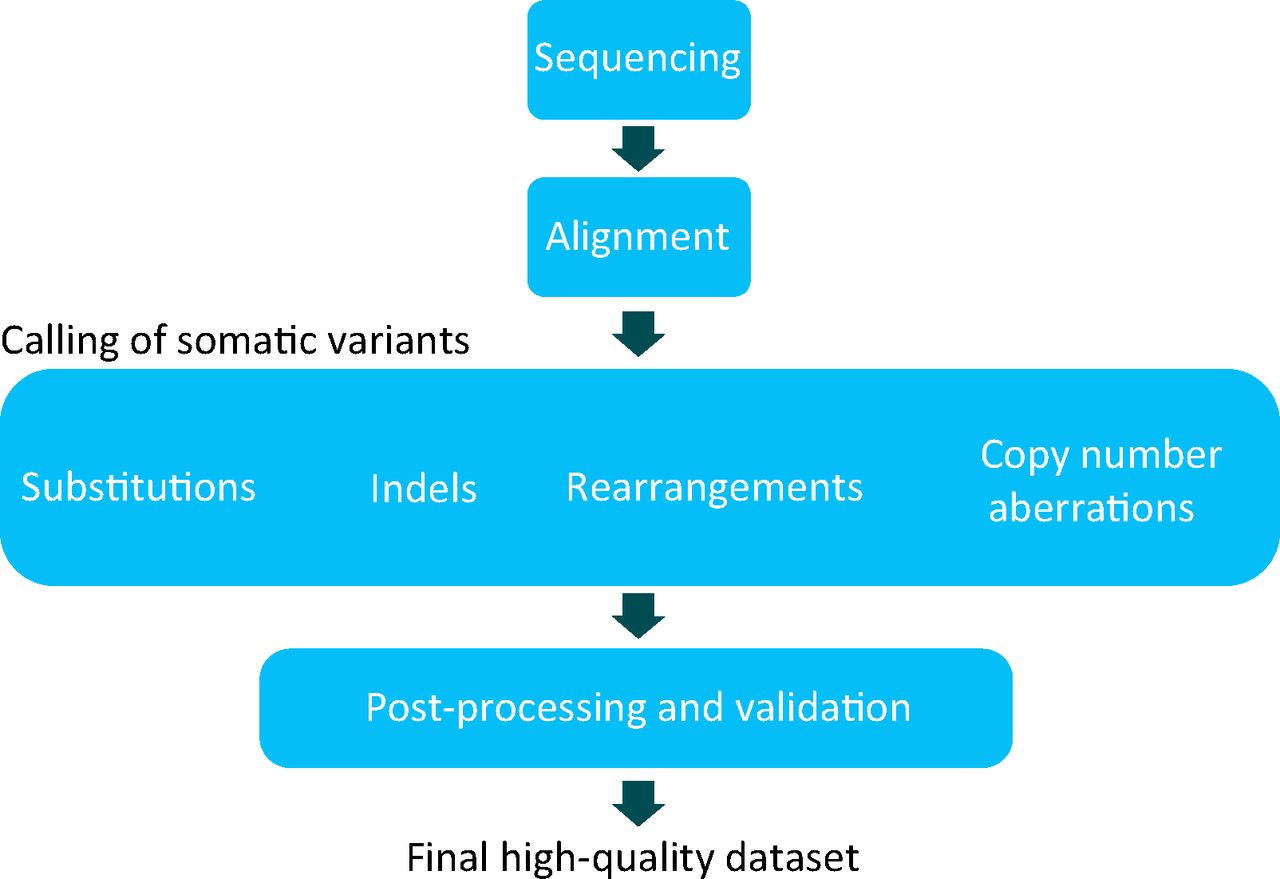
The process described here cannot be achieved without bioinformatic approaches, that is, a set of algorithms that help to perform alignment9 and mutation calling in a fast and efficient manner (Fig 3) (myriad different software packages have been developed to accomplish these tasks). However, the processing of raw data, the generation of mutations and the curation of such raw data sets demands considerable cost and expertise (Fig 3).2 Furthermore, obtaining high-quality catalogues of somatic mutation from WGS experiments is more demanding than that of whole exome sequence or targeted resequencing experiments, troubled by low-complexity sequence of intergenic regions, a high degree of repetitive sequence and, in parts, a less well-characterised reference genome. Similar to any screening tool in medicine, calling mutations in cancer and in the germline is not binary; it is based on probabilistic estimates and carries a measure of sensitivity as well as a false positive rate. Notwithstanding these technical challenges, the advantages gained from large-scale sequencing efforts are huge and will be summarised in the next section.
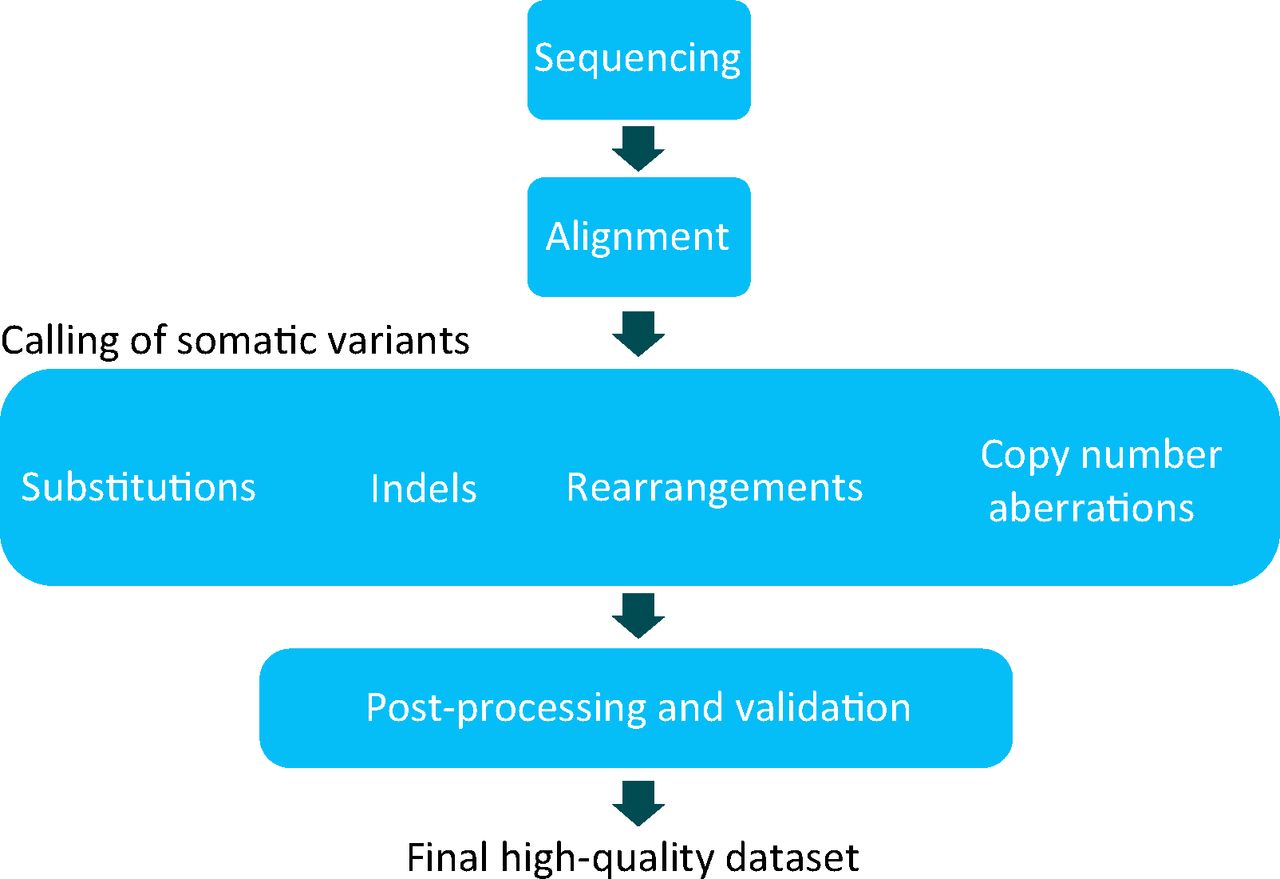
Bioinformatic processing. A schematic of a whole-genome sequencing and/or processing strategy for cancers. The two DNA samples obtained from each patient are processed into NGS libraries and sequenced independently. Raw sequences of the tumour and normal sample are aligned back to the reference genome. All classes of somatic mutation, including substitutions, insertions and/or deletions, somatic rearrangements and copy number aberrations, are sought using a range of bioinformatic tools. A high-quality data set requires further filtering or post-processing and might require validation or resequencing preferably on an alternative platform, of a subset of somatic mutations. Therefore, the final data set used for all downstream analyses is a highly curated data set. Note that exome and targeted sequencing strategies are not compatible with detection of all types of somatic mutation. NGS = next-generation sequencing.
What can be read from a cancer?
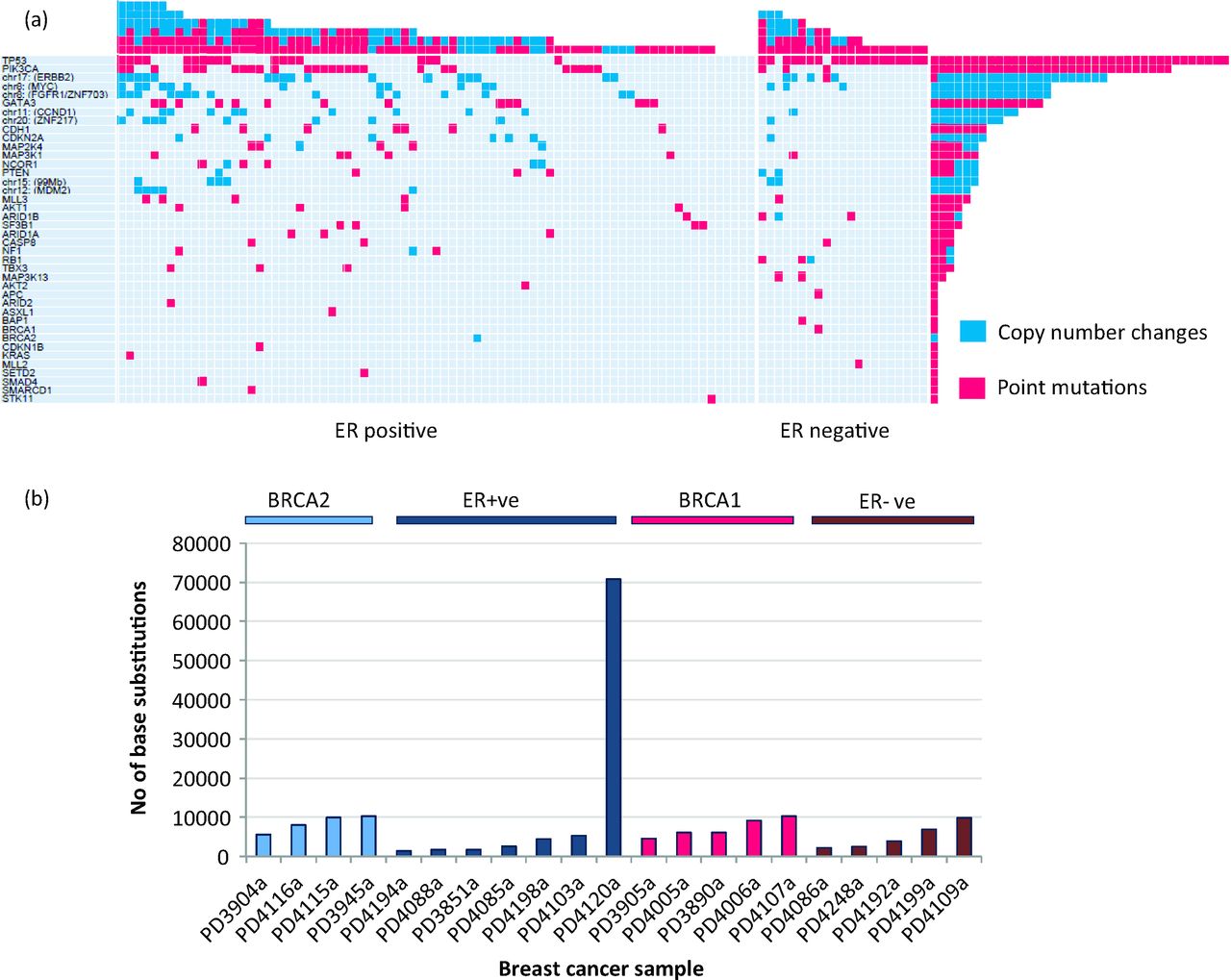
Decades of cancer research have been largely focused on the discovery of driver mutations in cancer genes, causally implicated in oncogenesis, because these become targets for developing new therapeutic agents.1 Here, the first key contribution of modern large-sale sequencing approaches, particularly exome and targeted-sequencing strategies, is the marked acceleration of the discovery of new cancer genes in recent years.10 Additionally, the affordability of sequencing today has resulted in more cancers being sequenced per experiment. Thus, rare low-frequency cancer genes present in common cancers,10–18 as well as common cancer genes present in rare cancers,19,20 are also increasingly being identified. These forays into identification of cancer genes demonstrates one startling point: that an enormous amount of intertumour heterogeneity exists. In an experiment of 100 exome-sequenced breast cancers, for example, no two individuals shared the same set of driver mutations11 (Fig 4).

Cancer genes and intertumour heterogeneity. (a) This image is taken from Stephens et al.11 and depicts the complexity and marked intertumour heterogeneity of cancer genes observed among breast cancers alone. These results were obtained from one large-scale NGS experiment of 100 breast cancer exomes. Each of the 40 cancer genes mutated in this experiment are documented on the left. The number of mutations in each gene in the 100 tumours is shown (rows), as is the number of driver mutations in each breast cancer (columns). Point mutations and copy number changes are coloured pink and blue, respectively. (b) In a separate whole-genome sequencing experiment involving just 21 different breast cancers, simply taking the total number of base substitutions into consideration, the degree of intertumour heterogeneity observed is marked, even within specific breast cancer subtypes. NGS = next-generation sequencing.
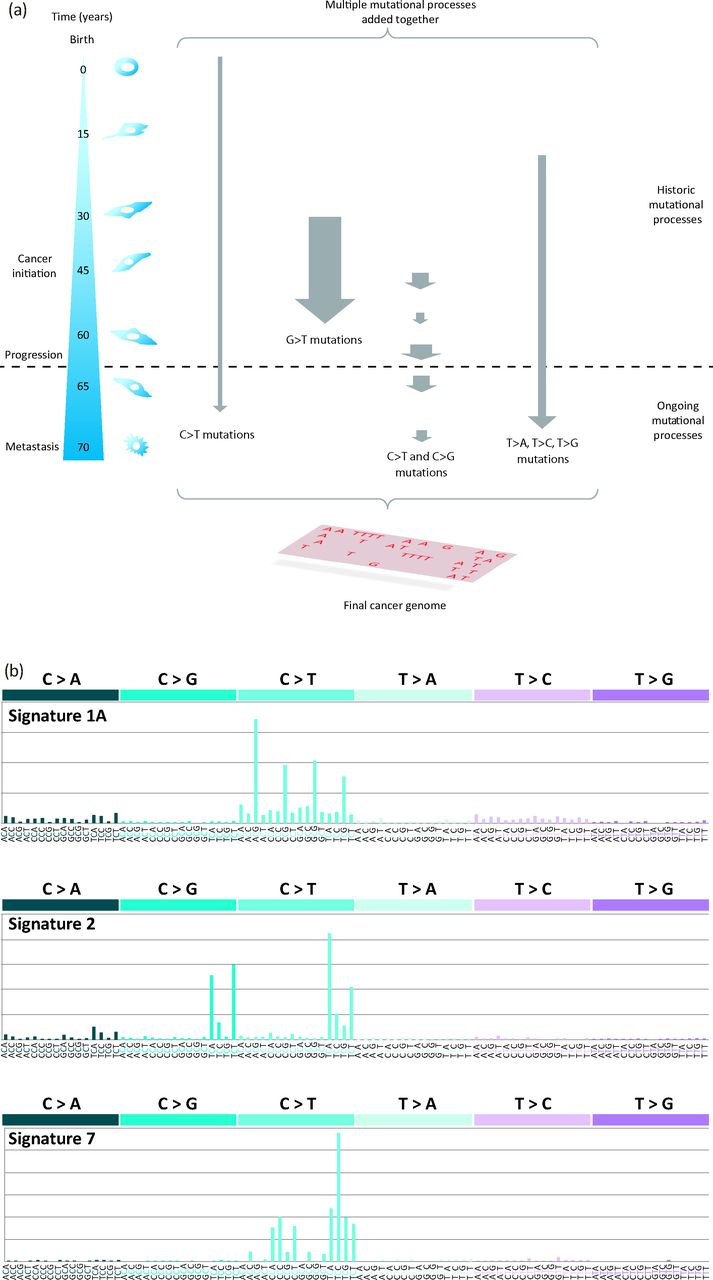
However, a cancer contains more than a mere handful of driver mutations. Each cancer bears many thousands of passenger mutations that might not be causative of cancer development but are a rich source of historical information.1,2 At the point of a patient's cancer diagnosis, the set of somatic mutations, whether base substitution, insertion and/or deletion of structural variation, that is revealed through sequencing of the cancer is the aggregate outcome of one or more biological perturbations or mutational processes. Each process leaves its own mark, its characteristic imprint or mutational signature on the cancer genome, defined by the mechanisms of DNA damage and DNA repair that comprise it (Fig 5).1,2 Whatever the nature of the mutagenic or repair mechanisms in operation, the final catalogue of mutations is also determined by the strength and duration of exposure to each mutational process (Fig 5).1,2 Some exposures might be weak or moderate in intensity, whereas others might be strong in their assertion. Similarly, some exposures might be on-going through the entire lifetime of the patient, even preceding the formation of the cancer, and some might start late or become dominant later in tumourigenesis (Fig 5).21
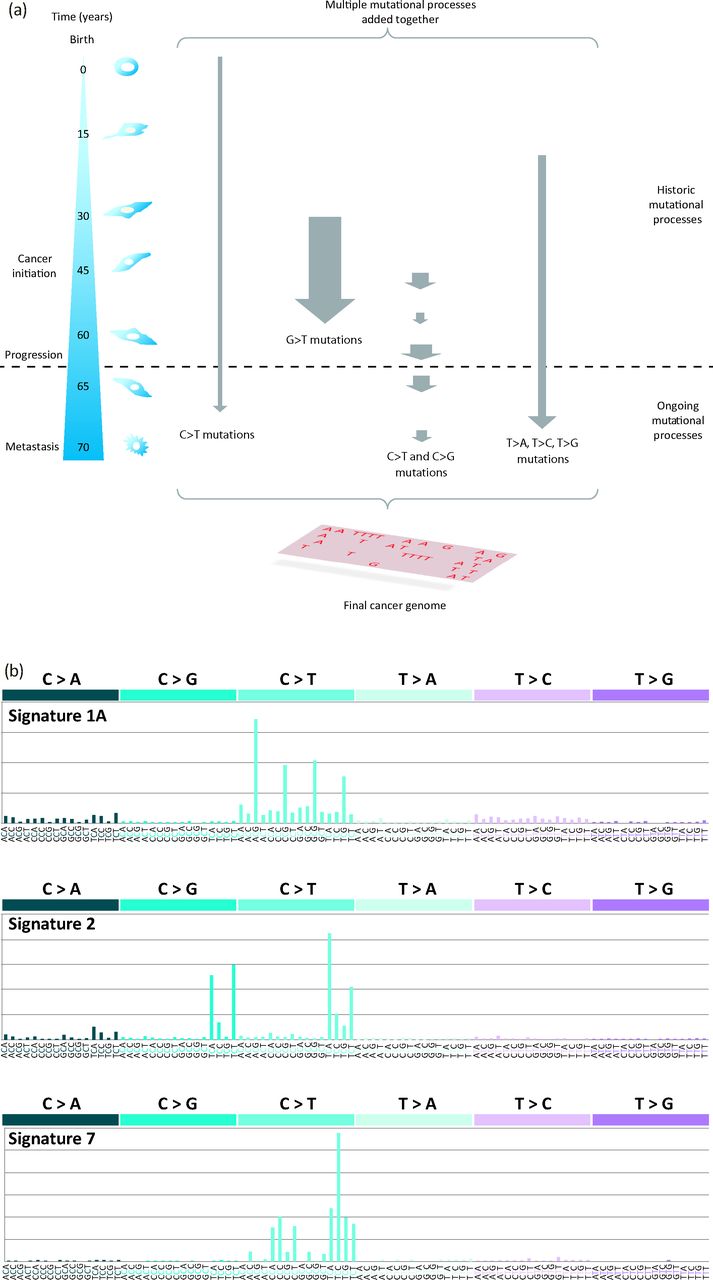
Mutational signatures in cancer genomes. (a) From the time of the fertilised egg through to the development of an invasive cancer, multiple mutational processes are likely to be operative, with each producing its own characteristic signature. At the point of diagnosis and of sequencing the cancer genome, the final mutation spectrum is a composite of the multiple mutational processes that have been operative that might show variation in the intensity (size of arrow) and duration (length of arrow) of exposure to each mutational process. This image is reproduced from Helleday et al.22 (b) Examples of mutation signatures extracted using mathematical approaches. Each signature comprises a 96-element pattern: six main substitution classes (C>A, C>G, C>T, T>A, T>C, T>G) that also takes the immediate flanking nucleotides into account (four possible bases 5′ and four possible bases 3′ to each mutated base; therefore, there are 16 possible options for each of the six substitution classes) giving 96 elements. Signature 1A, which is characterised by a large number of C>T mutations at a NpCpG trinucleotide pattern, is a ubiquitous signature identified in nearly all cancer types. It is believed to be the signature of deamination of methylated cytosines. Signature 2 is another common signature characterised by an excess of C>T and C>G mutations at a TpCpN trinucleotide. Both Signature 1A and Signature 2 are believed to be endogenous in origin. Signature 7 is characterised by an excess of C>T mutations at a CpCpN and a TpCpN sequence context. This signature is associated with exposure to ultraviolet radiation and is typically seen in malignant melanomas and other skin cancers. This image is reproduced from Alexandrov et al.23
WGS experiments result in vast data sets, characteristically thousands of mutations per WGS cancer (Fig 4b). The scale of such large data sets demands mathematical methods to distil the biological insights buried within.24 Intriguingly, such approaches have been used to unearth at least 21 different mutation signatures across 30 different cancer types,3 including signatures associated with past exposure to carcinogens, such as tobacco smoke in lung cancer and ultraviolet radiation in malignant melanoma.3 Apart from these known environmental exposures, endogenous enzymes that underlie mutagenesis, perhaps through normal physiological processes, have also been highlighted, including the ubiquitous deamination at methylated cytosines seen in nearly all human cancers2,3 and the activity of activation-induced cytidine deaminase (AICDA), which has a role in generating somatic hypermutation at immunoglobulin loci,3 in cancers of immune cells. However, many novel signatures have additionally been uncovered3 and the race is on to understand what causes these mutation signatures in cancer. Thus, the second fascinating insight permitted by modern sequencing approaches results from the ability to visualise and quantify mutation signatures from the totality of large mutation data sets obtained from cancer genomes.
Third, the digital nature of NGS readouts lends itself to mathematical modelling of other interesting and biologically pertinent features to the clinician, such as estimation of subpopulations of cells within a cancer.21,25 For example, coverage of 40-fold would mean that sequencing information from 40 DNA molecules is available at a particular genomic coordinate. A heterozygous variant in the germline would be expected to be present in approximately 50% of reads for a diploid genome (Fig 6) and a homozygous variant should be present in 100% of reads. In a tumour sample, a fraction of reads are likely to represent DNA from normal cells (from lymphocytes or stromal tissue contaminating the tumour sample), but the remaining reads should represent the tumour. A heterozygous variant in a diploid region in the tumour genome should be present in half of the remaining reads (Fig 6). If groups of mutations are found not to abide by this rule and instead be present in just a subset of the expected fraction of reads, then this can be used to infer the presence of a subclonal population in the cancer. Mathematical methods have been developed to identify such subpopulations and phylogenetic trees of each primary cancer can be constructed to a finite level of resolution.21 Cancer evolution does not have to be restricted to a primary tumour. Currently, efforts are being made to sequence cancers from one person that are separated either geographically (multifocal tumours or metastases) or temporally (recurrence). This results in further understanding of the relatedness of two tumour foci or how related a metastasis might be to its primary tumour.

Utilising the digital nature of NGS data to discern subclonal populations in a cancer. (a) Blue and purple reads joined by a dotted line represent forward and reverse reads, respectively of a 500 bp fragment. A higher resolution depiction of a section of a germline sample shows a 30-fold coverage of reads in the region of interest. The red marks represent a variant allele that is different to the reference genome. This heterozygous SNP in the diploid germline genome is seen in approximately 50% of reads or has a variant allele fraction of 0.5. This higher resolution schematic of a tumour sample also has 30-fold coverage but has 1/3 of reads originating from contaminating normal cells. In this region, which is diploid in the tumour, the somatic variant is a -heterozygous mutation and is present at a lower variant allele fraction (when compared with the germline genome) of 0.33. However, if the variant allele fraction of a true variant is lower than expected for the level of ploidy and normal contamination, when occurring in clusters, this might be taken as evidence of a somatic mutation in a subclonal population (intratumoural heterogeneity). By contrast, a polyploid region where a somatic variant is present on only one of multiple alleles will be present at a much lower variant allele fraction. (b) This image is reproduced from Nik-Zainal et al.21. This phylogenetic tree of a primary cancer was constructed by inferring the presence subclonal populations of base substitution mutations as well as subclonal copy number aberrations. Integration of these data sets enabled construction of such trees. To a finite level of resolution, it is possible to work out which mutations happen early or late during the evolution of this cancer. bp = base pairs; chr = chromosome; NGS = next-generation sequencing; SNP = single nucleotide polymorphism.
Significantly, modern sequencing technologies offer cancer medicine more than just the opportunity to identify cancer genes, to extract and quantify mutation signatures, and to describe the evolutionary history of a tumour. These new developments, although each remarkable in its own right, drive home a fundamental fact: that an extraordinary degree of tumour heterogeneity exists between patients. This cannot possibly be managed effectively with a one-size-fits-all chemotherapeutic approach. Instead, would it be possible to look forward to a future where molecular cancer genomic profiling could take the format of individualised reports with tailored therapeutic strategies to improve individual management?
What the future holds
I believe that the ability to provide an exhaustive, individualised genomic profile of each person's cancer is not far away. The cancer genomic report of tomorrow should not simply contain a record of the causative driver mutations in a patient's cancer. It should be a comprehensive profile of all mutation types, contain an interpretation of mutation signatures present in the primary tumour, and should suggest therapeutically relevant treatment strategies for each individual patient.
However, there are hurdles to negotiate before we will be able to achieve the stratified medicine cancer genomic report described above. First, the next generation of molecular genomic interpreters, whether they are molecular pathologists, geneticists or a new breed of cancer experts, need to be trained from today. The ability to read cancer genomes will need to reach the level of service delivery to be truly useful. This includes building the infrastructure to support this brand of clinicians: computational support, standard operating procedures for data handling and analysis, statistical and academic frameworks to operate from and legal and/or ethical guidelines, to name a few areas of development. Second, clinical trials of chemotherapeutic agents that incorporate improved genomic profiling of tumours are required. This is not a trivial exercise and years of work are still required before we will be in a position to match therapies to genomic status more effectively in the future. To reap the rewards from the technological advancement of sequencing tomorrow, we have to take action today.
Acknowledgements
SN-Z is a Wellcome–Beit Memorial Fellow 2013 and is personally funded through a Wellcome Trust Intermediate Clinical Research Fellowship (WT100183MA).
- © 2014 Royal College of Physicians
References








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.