
ABSTRACT
Kidney transplants do not last for the natural lifespan of most recipients. Of the reasons why transplants fail, damage by the immune system is the commonest cause. Understanding how the immune system recognises transplanted organs has increased significantly in recent years, but there is little insight into how organs are damaged, and no still no way of suppressing immune-mediated damage without exposing patients to the detrimental effects of long-term immunosuppression. In this article, we review the role of antibodies and B cells in immune-mediated damage of kidney transplants, and discuss the potential for manipulation of B cells to improve clinical outcomes.
Introduction
Kidney transplantation is the gold standard of treatment for patients with kidney failure, with better patient survival and quality of life compared with being on dialysis. However, many patients lose graft function during their lifetime and eventually return to dialysis. Up to 5% of the transplant population lose their graft in this manner each year, or approximately 700 patients in the UK. The most common cause for this premature graft failure is damage by the immune system. In order to address this problem, we need a better understanding of the immunological mechanisms that cause chronic damage.
Alloantibodies and graft loss
Although the pathogenesis of chronic graft dysfunction is multifactorial,1 immunological mechanisms are the major underlying problem in most kidney transplant recipients. There is now widespread recognition of the characteristic features of immune-mediated pathology seen on the transplant biopsy in most of these cases (Table 1). These features are strongly associated with the development of antibodies (Ab) directed against donor antigens (donor-specific Ab (DSA)), most commonly directed against human leukocyte antigens (HLA). These Ab, present in 20% of transplant patients, indicate that the recipient's immune system has been sensitised to HLA.
Diagnostic features of chronic, active AMR according to the most recent Banff 2013 classification.
Both DSA and HLA-specific Ab (HLA Ab) are useful predictive biomarkers for an increased risk of graft failure. Graft failure rates 5 years after testing are under 20% in patients without HLA Ab, compared to approximately 50% in those with DSA and 30–40% in patients with HLA Ab without DSA.2 The DSA or HLA Ab can appear many years before graft failure, during which time most patients develop graft dysfunction, manifesting most commonly as a slowly rising creatinine level and the development of proteinuria. A vast amount of experimental evidence supports the concept that these Ab are directly injurious to the graft, once bound to donor endothelium, via both complement-dependent and complement-independent mechanisms. Not surprisingly, complement-fixing Ab are associated with higher rates of acute and chronic rejection3 and are associated with positive C4d staining (a by-product of the activation of complement) on biopsies.4 These combinations of Ab, C4d and histopathological and/or molecular evidence of endothelial injury are now used together to diagnose ‘antibody-mediated rejection’ (AMR),5 a terminology reflecting the causative role that Ab are thought to play. However, while Abs certainly cause acute AMR, the evidence that they are the sole cause of ‘chronic AMR’ is substantially weaker.
AlloAb and graft survival
Approximately half of cases of chronic AMR diagnosed on transplant biopsy are negative for C4d and HLA Ab6 and acute rejection frequently precedes the development of DSA,7 both of which are consistent with the concept that other mechanisms contribute to graft dysfunction. Furthermore, many grafts continue to have stable function despite the presence of DSA/HLA Ab, consistent with the concept that Ab alone is insufficient in many cases to cause transplant dysfunction.
Another way of thinking of this is that some transplanted kidneys become resistant to AMR. This is called ‘transplant accommodation’ and was originally described in ABO-incompatible (ABOi) transplant recipients,8 but has also been seen in response to HLA-Ab.9 It is rarely described in non-ABOi recipients, but as a concept it may be relevant when trying to understand how patients with DSA or HLA Ab, particularly those with low-titre Ab, maintain good transplant function; some investigators have proposed that it may be more widespread than acknowledged.10
In chronic AMR, Ab alone are insufficient and cellular mechanisms may play an important role
In animal models of transplantation, alloAb alone are frequently insufficient to induce AMR, particularly in the absence of T and B cells, and in some rodent models, transfer of hyperimmune serum can even enhance graft survival. In rodent models of chronic AMR, transplant arteriosclerosis, the classical lesion of chronic rejection, can be mediated independently by any one of three mechanisms, including DSA, T cells and natural killer cells.11,12
T cells recognise graft major histocompatibility complex (MHC) antigens either via the ‘direct’ pathway (where intact MHC is recognised on donor cells) or the ‘indirect’ pathway (where CD4+ T cells recognise processed alloantigen presented as a peptide within self-MHC molecules on recipient antigen-presenting cells). T cells capable of indirect allorecognition persist for many years post-transplantation and are able to orchestrate rejection via several mechanisms including production of cytokines such as interferon (IFN)γ by monocytes/macrophages in delayed-type hypersensitivity responses; promotion of cytotoxic CD4+ and CD8+ T cells; and through production of Ab via interactions with B cells.13 Detectable ‘indirect’ CD4+ T cell anti-donor reactivity has been associated with chronic graft dysfunction in human cardiac14 and renal transplant patients,15 whereas quiescence has been associated with graft stability.16
The role of B lymphocytes in transplantation – lessons from autoimmune disease
The production of Ab requires a reciprocal interaction between antigen-specific CD4+ T cells recognising processed antigen and HLA on antigen-specific B cells. Thus, in transplant patients with HLA-specific DSA, the presence of DSA is proof of previous interactions between CD4+ T cells recognising donor HLA via the indirect pathway on donor-HLA-specific recipient B cells. As mentioned above, these ‘indirect’ T cells can mediate rejection via several effector pathways, but an important question is whether continuous interaction with antigen-specific memory B cells is involved in the maintenance of these effector responses.
Memory B cells play a key role in the development of T cell memory, as they are highly efficient at stimulating T-cell clonal expansion and proliferation, effective at up to 104 times lower concentrations of antigen than either macrophages or non-specific B cells; see Lanzavecchia et al.17 In contrast to naÔve and activated B cells, their survival is not dependent on the continued presence of antigen, and they have a lower threshold for activation. On re-exposure to antigen they rapidly expand and differentiate into short-lived antibody-producing plasma cells, leading to a burst of Ab production. Memory B cells survive long-term, and differ significantly from plasma cells in that they reside in lymphoid tissue and do not require B cell activating factor (BAFF) or a proliferation inducing ligand (APRIL) survival factors. B cell memory is maintained by the survival of these antigen-specific cells, even when Ab levels are undetectable or low.
B-cell depletion using the anti-CD20 monoclonal antibody, rituximab, has been remarkably effective in treating patients with autoimmune disease, improving clinical and radiological outcomes in rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and multiple sclerosis (MS). The mechanism by which this occurs is incompletely understood, but one model is that these diseases arise due to a loss of tolerance to self-antigens, leading to a ‘vicious cycle’ of presentation of autoantigen by B cells to CD4+ T cells and resulting in reciprocal expansion of T- and B-cell effector and memory responses, which includes differentiation into plasma cells and autoantibody production.18 Rituximab, by depleting B cells, breaks this cycle, as evidenced by the accompanying reductions in T- and B-cell numbers in joint (in RA) or cerebrospinal fluid (in MS).
The effectiveness of rituximab highlights the crucial role that memory B cells play in sustaining effector mechanisms in these diseases. This is supported by some animal models of autoimmunity, in which tolerance is broken by vaccination with autoantigen, following which B-cell depletion, performed after the onset of disease, significantly reduces disease severity.19 Paradoxically, if B-cell depletion is performed prior to the initiation of disease in these animal models, disease severity is increased, suggesting that B cells play a role in sustaining tolerance to self.20 This is consistent with studies in which adoptive transfer of interleukin (IL)-10-producing B cells reduces disease severity in mouse models of MS and RA, possibly through the promotion of regulatory CD4+ T cells,18,21 and with clinical observations after rituximab administration to patients with autoimmune disease. A feature of prolonged remission in these patients is a loss of memory B-cell subsets, and the return of immature, transitional B cells, which are thought to re-establish tolerance by virtue of their predominant IL-10 secretion and regulatory properties. Indeed, there is some evidence that these types of B cells are dysfunctional in patients with SLE, suggesting they may be involved in the initial loss of self-tolerance.22
In the transplantation arena, B cells have also been associated with operational tolerance and graft stability,23,24 suggesting that regulatory B cells may play a role in controlling alloimmunity. Recently, Nouel et al reported a reduction in the number and functional activity of transitional B cells in patients with chronic AMR,25 and Cherukuri et al reported associations between chronic rejection and a shift towards pro-inflammatory cytokine secretion by transitional B cells, compared with the predominant IL-10 secretion seen by transitional B cells in patients with stable graft function.26
Optimising graft survival through B-cell depletion
Highly sensitised transplant recipients, ie those with HLA Ab, are at the greatest risk of acute and chronic rejection. All these patients have memory B and T cells specific for allogeneic HLA, akin to patients with autoimmune disease who have memory cells specific for autoantigens. B-cell depletion improves graft outcomes after transplantation into sensitised animals by lowering Ab levels and impairing T-cell activation and proliferation, reducing rates of acute cellular rejection and delaying or inhibiting development of chronic AMR.27 Rituximab has also been successfully used in induction protocols for highly sensitised transplant recipients (in sensitised recipients, this is not the same as deleting B cells prior to disease induction in immunologically-naÔve animals) and is also frequently used in combination with intravenous immunoglobulins for the rescue treatment of acute AMR.28 Studies have been limited by small patient numbers and uncontrolled design, although a recent randomised controlled trial comparing IVIG + placebo and IVIG + rituximab for induction therapy of highly-sensitised patients reported a significant benefit from rituximab treatment, and was discontinued early due to a high rate of severe acute AMR in the placebo arm.29
Its role in the treatment of chronic AMR remains unclear, with small numbers of patients reported as benefiting from B cell-depleting treatment in uncontrolled studies.30 The focus of our work has been trying to understand how B cells interact with T cells in patients with chronic AMR, diagnosed on the basis of either protocol or ‘for cause’ transplant biopsies. In approximately 20% of patients, we have demonstrated detectable IFNγ production (using Enzyme Linked ImmunoSPOT assay) by CD4+ T cells activated via indirect allorecognition, indicating that B cells are important for processing and presenting donor antigens. In a further 20%, this B-dependent reactivity is only revealed by depletion of CD25+ T lymphocytes and these cells have a putative regulatory phenotype by flow cytometry. Finally, in another 20%, IFNγ production is revealed by depletion of CD19+ cells, suggesting that B cells in these individuals are behaving as functional suppressor cells, a finding supported by preliminary analysis of how B cells from these individuals behave after polyclonal stimulation (they tend to make IL-10). Interestingly, we have not been able to find any correlations between this ELISPOT pattern and either the number or phenotype of transitional B cells in these patients. Our provisional conclusions from all this work are that B cells play a significant role in indirect alloantigen-specific donor T-cell reactivity in some patients with chronic AMR, but there is active suppression of T-cell immunity by distinct phenotypes of T and B cells in approximately 40% of patients, suggesting a complex heterogeneity of immune-mediated graft dysfunction in these patients (Fig 1).

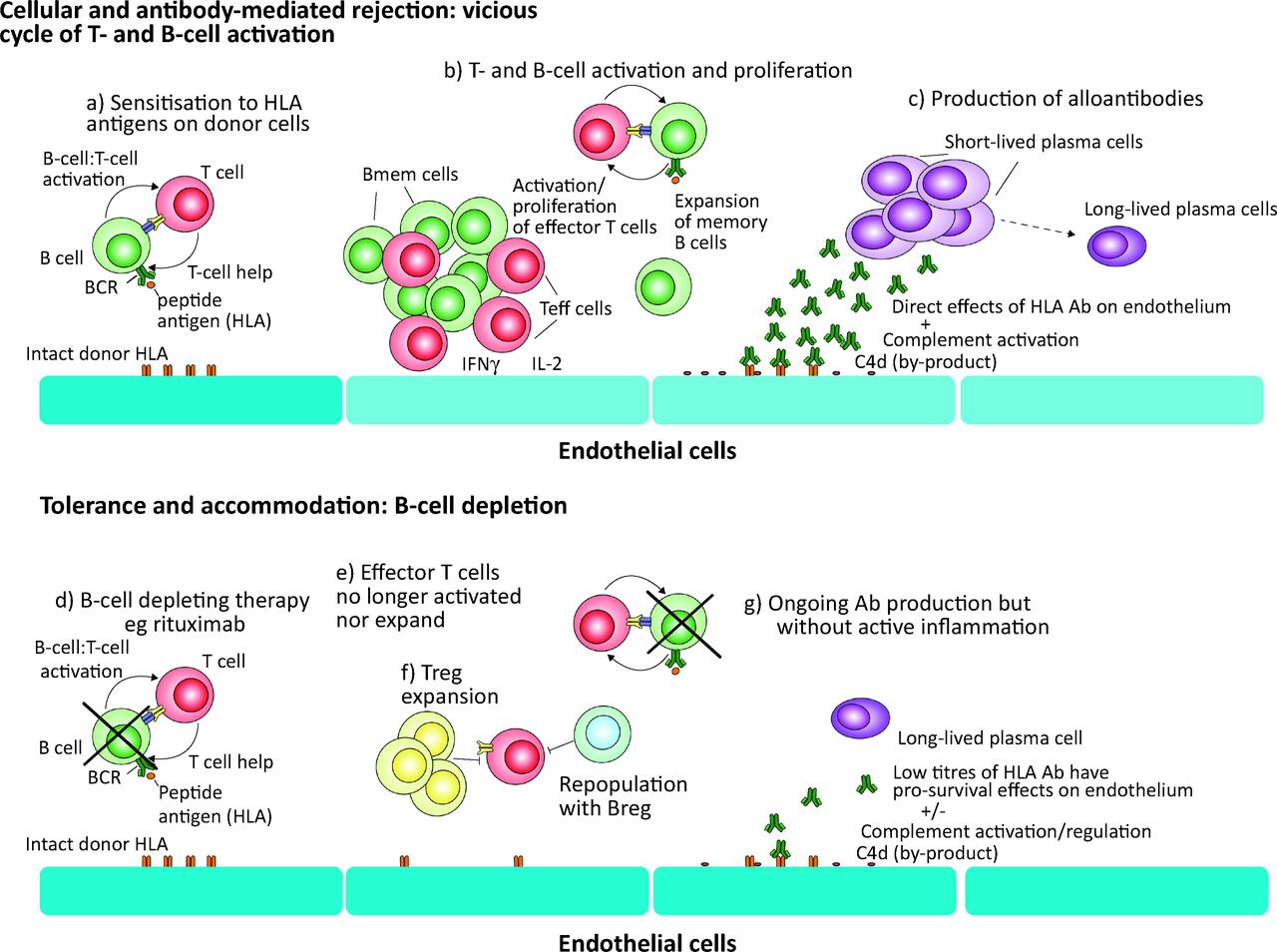
The role of B cells in driving graft rejection in an Ab-independent manner and the potential benefit of targeting B cells. The pathogenesis of chronic antibody-mediated rejection is not fully understood, and the widely-held conception that donor-specific Ab, often specific for HLA, underlie the graft damage, is likely to be oversimplistic. Based on the available data, and experience from the effects of treating autoimmune disease with B-cell depletion, we suggest a model in which (a) sensitisation of T and B cells to graft antigens leads to a vicious cycle of (b) activation and expansion of both Teff and Bmem. (c) CD4+ effector T cells produce high levels of pro-inflammatory cytokines including IFNγ and IL-2, in a delayed-type hypersensitivity response, which can induce direct damage. Simultaneously, there is a rapid expansion of short-lived plasma cells that may produce a burst of high levels of Ab directed at HLA, which can cause direct damage to endothelial cells as well as indirect effects through complement activation and antibody-dependent cell-mediated cytotoxicity. (d) When B-cell-depleting therapy is administered this vicious cycle is broken. (e) The activation of T cells is impaired, with reductions in the number and function of memory T cells. (f) This environment may promote the development of Tregs, and as transitional and naÔve B cells repopulate from the bone marrow these are likely to further promote the development of tolerance to donor antigens, as well as cellular regulatory pathways. (g) Ab production may continue, as long-lived plasma cells can reside for many years, but in the absence of activated B cells contributing to a plasma cell pool, this is likely to be at low levels that can further promote the development of accommodation. Ab = antibody; BCR = B-cell receptor; Bmem = memory B cells; HLA = human leukocyte antigen; IFN = interferon; IL = interleukin; Teff = T effector cells; Treg = regulatory T cells.
These data suggest that rituximab might be effective at disrupting on-going cellular immune responses in some individuals with chronic AMR, but might have adverse effects in others (ie those with putative regulatory B cells suppressing alloreactivity). Therefore, to address the role of rituximab in chronic AMR, we are conducting a randomised, controlled trial (RituxiCAN-C4 clinicaltrials.gov identifier NCT00476164). The objective is to test whether rituximab can stabilise kidney function (reduce ΔeGFR) in patients with biopsy-proven chronic AMR who fail to respond to optimised oral immunosuppressive therapy. After a run-in period on optimised oral therapy, 30 patients who fail to respond will be randomly allocated to rituximab or control (no rituximab) arms. Those responding to optimised oral immunosuppression (or not consenting to randomisation) enter a three-year observational phase to inform the natural history of chronic AMR. To date, 58 have been recruited and 20 randomised. More than 30 have completed three years of follow-up or suffered graft failure. The trial is based on an estimate that 11/15 patients given rituximab and 4/15 controls will stabilise within 5 months of randomisation. An interim analysis of outcomes will be performed in late 2014.
Conclusion
The production of Ab is a product of a reciprocal interaction between T and B cells that involves important ‘cross-talk’ through which the T cells provide crucial activation signals for the B cells, which may either become Ab-producing cells or antigen-specific memory B cells. Antigen-specific B cells provide potent activation and proliferation signals to T cells, thus leading to T cell effector mechanisms which can themselves cause injury, and potentially drive graft rejection. These immune effector processes underpin the damage that occurs in transplanted kidneys suffering from chronic AMR, which has emerged as the commonest cause of premature allograft failure. The critical questions we are asking relate to the importance of the cellular effector responses, compared with Ab, and whether targeting memory B cells, using rituximab, can prevent the progressive functional decline often seen in these patients. Improved understanding of the precise mechanisms involved in chronic AMR, and the ability to distinguish which mechanism is active in individual transplant recipients, should lead to the development of rational, safe and effective treatments to prolong graft survival.
Acknowledgements
The authors acknowledge that this research was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas’ NHS Foundation Trust and King's College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. The work referred to here was supported by MRC grants G84/6713 and G0801965, Roche Organ Transplant Research Foundation Award ROTRF 53331024, Novartis Pharmaceuticals non-promotional grant TRA10-087 and KRUK grant RP3/2011.
- © 2014 Royal College of Physicians
References








{kind=link}
Jump to section
- Article
- ABSTRACT
- Introduction
- Alloantibodies and graft loss
- AlloAb and graft survival
- In chronic AMR, Ab alone are insufficient and cellular mechanisms may play an important role
- The role of B lymphocytes in transplantation – lessons from autoimmune disease
- Optimising graft survival through B-cell depletion
- Conclusion
- Acknowledgements
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.