
ABSTRACT
Atypical haemolytic uraemic syndrome is a thrombotic microangiopathy with a predominant renal phenotype. Research developments in the last 15 years have led to the elucidation of the role of complement over activation in the pathogenesis of the disease. This was to lead to the successful introduction of targeted pharmacological therapy, in the form of the complement inhibitor, eculizumab.
Introduction
Haemolytic uraemic syndrome (HUS) is characterised by the clinical triad of thrombocytopenia, microangiopathic haemolytic anaemia (MAHA) and acute kidney injury (AKI). The typical form, which accounts for the majority of cases, is caused by enteric infection with Shiga-toxin (Stx)-producing bacteria, most commonly Escherichia coli (E coli) serotype O157:H7 and Shigella dysenteriae type 1.1 Any HUS not caused by Stx has been classified as atypical HUS (aHUS);2 it is rare (the true incidence is unknown but has been estimated to be around 2 per million)3 but is associated with significant morbidity and mortality. Research in the last 15 years has brought about major advances in our understanding of the pathogenesis of aHUS, now recognised in the majority of cases to result from complement dysregulation. This ultimately resulted in the introduction of complement modulatory therapy for aHUS, with a consequent improvement in patient outcome.
This review will focus on the role of the complement system in aHUS and the introduction of the complement inhibitor eculizumab.
The complement system
The complement system comprises over 30 proteins4 and has a number of key physiological functions: it protects the host from invading pathogens by stimulating the inflammatory response and eliminates pathogens through opsonisation and cell lysis; bridges the innate and adaptive immune systems; and allows disposal of immune complexes and injured or apoptotic host cells.4
Complement activation can be initiated by three different pathways: the classical, alternative and lectin pathways, which converge with the generation of the C3 convertase and share a common terminal pathway4 (Fig 1). Activation of the classical pathway is initiated by immunoglobulin and involves the pattern recognition molecule (PRM) C1q.4 The lectin pathway is activated when the PRMs mannose binding lectin and ficolin bind to carbohydrates on the surface of micro-organisms.4 In contrast, the alternative pathway is constitutively active, with C3 undergoing spontaneous hydrolysis in a tick-over mechanism, which results in the continuous deposition of C3b onto the surface of cells in the vicinity. On a foreign (activating) surface such as a bacterium, C3b binds to factor B (CFB), which is cleaved by factor D to form the C3 convertase (C3bBb) which subsequently cleaves more C3 and thereby amplifies the cascade4 (Fig 1). In the common terminal pathway the C3 convertase binds additional C3b to generate the C5 convertase which cleaves C5 to C5a, an anaphylatoxin, and to C5b, which initiates formation of the lytic membrane attack complex (C5b-9).4 A rapidly amplifying immune response, which evolved to destroy invading microorganisms, necessitates regulation in order that host cells are protected from collateral damage. This bystander damage is prevented by a number of fluid phase (eg complement factor H [CFH] and complement factor I [CFI]) and membrane-bound (eg membrane cofactor protein [CD46]) complement regulatory proteins4 (Fig 1).
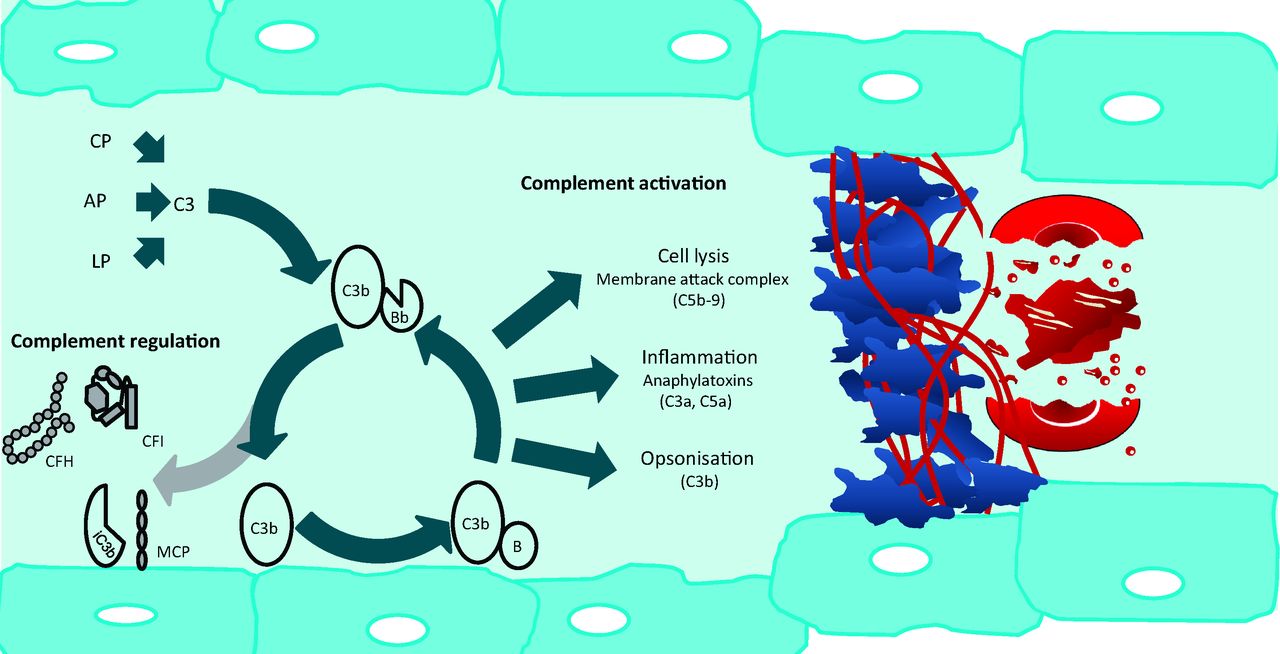
Complement activation and regulation in the pathogenesis of aHUS. The AP of complement constantly undergoes tick-over but can also be primed by the CP and LP. C3b interacts with factor B (B), which then is cleaved by factor D to form C3bBb. This enzyme complex is attached to the target covalently via C3b while Bb is the catalytic serine protease subunit. C3 is the substrate for this convertase, thus creating a powerful feedback loop. Unchecked, this leads to activation of the terminal complement pathway with generation of the effector molecules, the anaphylatoxin C5a, and the membrane attack complex (C5b-9). Host cells are protected from bystander damage by downregulation of the AP by complement regulators including CFH, CFI, and MCP. Activating mutations in C3 and CFB and loss-of-function mutations in CFH, CFI, and MCP, in addition to autoantibodies to CFH and CFI, result in overactivation of the AP with resultant aHUS. Glomerular capillary wall thickening is seen due to endothelial cell swelling and accumulation of material between the endothelial cell and the basement membrane. This narrowing of the vessel lumen, in addition to the platelet and fibrin thrombi, results in occlusion of the glomerular capillaries. This results in the microangiopathic haemolytic anaemia and acute kidney injury. AP = alternative pathway; aHUS = atypical haemolytic uraemic syndrome; C3bBb = AP C3 convertase; CFH = complement factor H; CFI = complement factor I; CP = classical pathway; LP = lectin pathway; MCP = membrane cofactor protein.
aHUS manifests when inherited and/or acquired defects in the complement system result in an imbalance between complement activation and regulation in the glomerular vasculature.2
Recognition and diagnosis of aHUS
Diagnosis
aHUS belongs to a group of conditions called thrombotic microangiopathies (TMAs), which are characterised by endothelial injuries that result in occlusive intravascular thrombi in small vessels, with resultant MAHA. Elevated creatinine, lactate dehydrogenase and bilirubin with a low haptoglobin, in addition to a Coombs’-test-negative haemolytic anaemia, and thrombocytopenia are all consistent with a diagnosis of HUS. A normal coagulation profile will exclude disseminated intravascular coagulation (DIC).
Upon identifying a TMA, three major differential diagnoses need to be considered: Stx-HUS, thrombotic thrombocytopenic purpura (TTP) and aHUS. Stx-HUS can occur in the context of outbreaks of Stx-producing E coli, or individually. Cases should be confirmed or excluded on the basis of stool culture and Stx-PCR (Table 1).
Investigating thrombotic microangiopathies.
TTP occurs when deficiency in a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13), an enzyme that usually cleaves von Willebrand factor (vWF), results in unusually large vWF polymers that trigger platelet aggregation.5 ADAMTS13 activity measurement must be performed on a blood sample taken prior to commencing plasma exchange (PEX), with a level <10% diagnostic of TTP.5 It has been suggested that TTP and aHUS can be presumptively distinguished on the basis of clinical evidence prior to the availability of the ADAMTS13 result, with a serum creatinine of >150–200 μmol/l or a platelet count >30,000 mm3, virtually excluding a diagnosis of severe ADAMTS13 deficiency.6
Complement analysis in cases of aHUS should include serum levels of C3, C4, CFH, and CFI. Low C3 levels are seen commonly in patients with mutations in complement genes and may point to a complement-mediated process. However, normal C3 levels do not exclude the presence of a mutation in, or autoantibodies against, the complement system.2 A full biochemical and genetic analysis of the complement system is warranted in aHUS (Table 2).
Investigating aHUS.
Other causes of renal TMA which can be excluded with specific investigations include the antiphospholipid syndrome, systemic lupus erythematosus, HIV, systemic sclerosis and cobalamin C disease2,5 (Table 1).
A renal biopsy is not always undertaken – the risk of bleeding complications must be considered – but histological evaluation in aHUS will demonstrate platelet and fibrin thrombi occluding glomerular capillaries and sometimes fibrinoid necrosis.2
Clinical presentation
Presenting symptoms in adults are sudden and include complications of AKI such as biochemical derangement, volume overload and hypertension. Extra-renal manifestations are reported in 10–20% of individuals with aHUS, with neurological symptoms being the most common and comprising a range of symptoms including irritability, seizures, visual abnormalities and coma.2,3 It may be difficult to differentiate between cerebral TMA and sequelae of severe hypertension, in which case brain magnetic resonance imaging can be helpful.2,3 Other extra-renal manifestations have been described, including myocardial infarction, digital ischaemic gangrene, ocular involvement, pancreatitis and pulmonary haemorrhage.2,7
Penetrance and triggering events
The age of onset of aHUS varies, ranging from the neonatal period to adulthood, with approximately 40% presenting for the first time in adulthood.3 In addition, incomplete penetrance has been reported for all of the genes associated with aHUS.2 Penetrance is influenced by genetic modifiers, such as common single nucleotide polymorphisms and mutations, as well as environmental modifiers.2 Environmental factors include infections, pregnancy, drugs and diarrhoeal illnesses, and are thought to precipitate aHUS by triggering the alternative complement pathway, which in genetically susceptible individuals cannot be regulated appropriately.2
Complement and aHUS
In patients with aHUS, loss-of-function mutations in complement regulators, activating mutations in complement components and autoantibodies to complement regulatory components have been reported.
Loss of function mutations
Complement factor H
CFH is the principal fluid-phase complement regulator. It functions by by competing with CFB for C3b binding, operating as a co-factor for CFI-mediated C3b proteolysis, and promoting C3 convertase decay.2,4 It can also bind to polyanions such as glycosaminoglycans, thus protecting host cell surfaces.8 Accounting for approximately 25% of the genetic predisposition to aHUS, mutations in CFH are the most frequently identified genetic abnormality in aHUS patients.2 They were first discovered in 1998 when a genetic linkage study was undertaken in three families with HUS.9 Most CFH mutations do not result in a quantitative deficiency of CFH, but give rise to proteins shown in functional analyses to be defective in their control of the alternative complement pathway.2,10
Complement factor I
CFI is a serum serine protease that cleaves C3b and C4b in the presence of its cofactors (eg CFH and CD46).2 Sanger sequencing of CFI in three individuals with aHUS led to the first report of the role of mutations in CFI in 2004.11 CFI mutations constitute around 5–10% of aHUS, and defective regulation of both the alternative and classical pathways has been demonstrated in functional analyses.12,13
Activating mutations
In recent years, activating mutations in C3 and CFB, complement components from which the amplifying C3 convertase is comprised, have been described. C3 mutations are found in around 2–10% of aHUS cases, whereas CFB mutations are rare.16,17 They result in increased C3 convertase activity and consequently greater complement mediated damage to glomerular endothelium.18
Autoantibodies
Acquired, as well as genetic, abnormalities have been described in the pathogenesis of aHUS. In 4–14% of aHUS patients (and reportedly a higher proportion of paediatric patients), autoantibodies to CFH have been identified.19,20 Autoantibodies to CFI have been reported but are rare. Functional studies have indicated a pathogenic role for CFH autoantibodies20 but there is no convincing evidence for direct pathogenicity with CFI autoantibodies.21
Clinical course and prognosis
Outcome data from historical series that precede the introduction of eculizumab showed a poor overall prognosis for aHUS. In a French cohort, mortality at one year was reported as 6.7% in children and 0.8% in adults; progression to end stage renal disease (ESRD) after the first episode was 16% in children and 46% in adults.22 Prognosis varies depending on the genotype. CD46 mutations are associated with the best outcomes; in the largest published cohort of genetic screening in aHUS patients, which looked at 273 patients at 3 years, no patient with a CD46 mutation had died, and only 6% had reached ESRD.23 In contrast, individuals with CFH, CFI and C3 mutations have much poorer outcomes. In the same cohort, mortality after 3 years was 23% for individuals with CFH mutations, and 53% had reached ESRD.23 The natural history of the disease is that relapses are common for those individuals who survive and do not reach ESRD, especially for those with CD46 mutations.22 In the French cohort, 35% of adults experienced at least one relapse (median follow up 57 months), with 85% of these occurring in the first year.22 However, in this new era of complement inhibitory therapy outcomes are expected to significantly improve.
Conventional management
Supportive management is important, and consideration should be given to transferring patients to a critical care or nephrology unit. Acute kidney injury and hypertension must be managed appropriately, and the proportion of adults who require renal replacement therapy during the first episode of aHUS has been reported at around 80%.22 Platelet transfusions can worsen the TMA and are not recommended.3
Prior to the introduction of eculizumab, national guidelines recommended that patients presenting with aHUS should be offered a trial of plasma exchange (PEX). There is no randomised controlled trial data to support the use of PEX, but on mechanistic grounds it is a logical treatment.24 PEX results in removal of autoantibodies and hyper-functional complement components, and replacement of defective complement components.2
The individual response to PEX is variable, with reported response rates of 30–70%.24 Genotype explains some of the variation; patients with abnormalities of the membrane-bound regulator CD46 will not respond to PEX as well as those patients with dysfunctional soluble complement components.24
The current recommendation for patients with suspected aHUS is to commence PEX if the ADAMTS13 activity is not immediately available, but if it is available and is not <10% then eculizumab is considered first line therapy.5
Management with eculizumab
Pharmacology
Eculizumab is a humanised recombinant monoclonal antibody that binds with high affinity to human complement component C56,7 and prevents cleavage of C5 into C5a and C5b, thereby inhibiting complement pro-inflammatory and lytic activity.6,7 The drug was designed to be minimally immunogenic, with a hybrid heavy chain comprising IgG2, which does not bind Fc receptors, and IgG4, which does not activate complement.
Eculizumab is administered intravenously, and achieves complete complement block-ade when its serum concentration reaches 35 μg/ml. It was first approved for treatment of paroxysmal nocturnal haemoglobinuria (PNH).6,7
Use of eculizumab in aHUS
The successful use of eculizumab in aHUS was first reported in two separate case reports published in 2009. More promising case reports followed, which led to two non-randomised prospective trials published in 2013.
The first trial included 17 patients with progressive TMA resistant to PEX, and the second included 20 patients with aHUS who were being maintained on long term PEX.25 In these trials eculizumab was shown to be effective in inducing haematological remission and was associated with significant time-dependent improvement in renal function.25 Eculizumab is now considered optimal treatment for aHUS.
Eculizumab in kidney transplantation
Historically, the overall outcome of renal transplantation in patients with aHUS is poor, largely because of recurrence in the allograft. In one cohort the mortality was 7% and graft failure was 50% after 5 years post-transplantation.26 Recurrence risk is predicted by genotype, with the highest risk associated with CFH mutations and the lowest with CD46 mutations, because allograft cells express functional CD46.26,27
For patients with defects in complement components that are synthesised in the liver, a combined liver–kidney transplant is a logical strategy which has been successful in conjunction with PEX, although mortality is higher than would be expected for kidney transplantation alone.5,27
Eculizumab has been successfully used prophylactically at the time of renal transplantation in individuals at high genetic risk of recurrence, and as rescue therapy for aHUS recurrence post-transplantation. Because of this, eculizumab is considered the gold standard in renal transplantation in aHUS patients.
Adverse effects of eculizumab
The membrane attack complex is key in the defence against encapsulated organisms, and patients with congenital C5 deficiency experience recurrent Neisseria meningitidis infections;6 this is therefore a fundamental concern in patients receiving eculizumab. In clinical trials of eculizumab in PNH, the Neisseria infection rate was 4.2 cases per 1,000 patient-years.6 Current practice is to administer the meningococcal vaccine and long-term prophylactic antibiotics to patients receiving eculizumab.5
The consequences of long-term eculizumab are not yet known. It has been reported that renal biopsies in patients on eculizumab have shown evidence of ongoing C3 deposition, as well as eculizumab deposition in the kidney.28
Economics of eculizumab
A barrier to the widespread use of eculizumab is the very high cost. NHS England's economic analysis suggests that the discounted incremental cost-effectiveness ratio for eculizumab, versus standard care, is around £521,000 per QALY gained for a 23-year-old cohort.29
Eculizumab treatment failure
With its increasing use in clinical practice, it has become clear that in rare cases disease will break through despite adequate eculizumab levels. In a Japanese PNH population, a single nucleotide polymorphism in C5 at amino acid R885 resulted in a failure of complement blockade.7 Additionally, in around 30% of familial aHUS cases no complement defect has been detected. Recently, mutations in diacylglycerol kinase-epsilon (DGKε) have been reported in aHUS.30 DGKε is part of an intracellular signalling cascade and although its role in the pathogenesis of aHUS has yet to be fully elucidated, it is not thought to participate in the complement system. In keeping with this, several individuals with mutations in DGKε have failed to respond to eculizumab.30
Unanswered questions
It is not yet clear what the optimal duration of eculizumab treatment is, and when – if at all – it might be appropriate to discontinue it. There are anecdotal reports of aHUS relapse when eculizumab was discontinued and when doses were delayed or missed.7,25 A trial of eculizumab withdrawal in aHUS is required.
Summary
In the last 15 years, it has become clear that overactivity of the alternative pathway of complement is central to the pathogenesis of aHUS. This had led to the introduction of a successful treatment in the form of the complement C5 inhibitor, eculizumab.
Conflict of interest
DK has received honoraria for consultancy work from Alexion Pharmaceuticals.
Acknowledgements
DK is a Wellcome Trust intermediate clinical fellow.
- © 2014 Royal College of Physicians
References








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.