
ABSTRACT
IgG4-related disease is a recently recognised multi-system disease. Common organ involvement includes the pancreas, biliary tree and salivary glands. Central nervous system involvement has been infrequently reported. In a single-centre cohort of 84 patients, we report cerebral involvement in three (4%) patients. Details of cerebral involvement in these patients are outlined, including pituitary involvement in two patients and a diffuse autoimmune-like encephalopathy in the other.
Introduction
IgG4-related disease (IgG4-RD) is an emerging disease entity that was initially described in 2003 as a ‘multi-systemic disease’ in patients with autoimmune pancreatitis (AIP).1,2 At presentation, over 50% of patients show evidence of multi-organ disease.3,4 IgG4-RD involvement has been reported in almost every organ system, including the kidneys, biliary tree, liver, salivary gland, orbit, breast, pericardium, aorta, skin, lungs and prostate.5–8 IgG4-RD is associated with significant patient morbidity and mortality.9
A number of diagnostic criteria have been used for IgG4-RD, including the recent International Consensus Diagnostic Criteria (ICDC).10 During the period of this study, the HISORt criteria proposed by the Mayo Clinic were used in the diagnosis of AIP11 and its extra-pancreatic manifestations. These criteria include diagnostic histology, characteristic imaging, elevated serum IgG4 levels on serological testing, other organ involvement and response to steroid therapy. The pathological hallmark of IgG4-RD is a lymphoplasmacytic tissue infiltration with IgG4-positive plasma cells (30–50 IgG4-positive cells per high-powered field), a storiform fibrosis (whorled or cartwheel appearance) and obliterative phlebitis.12 Tissue eosinophilia may also be present. Serum IgG4 levels are characteristically raised, but may be normal in up to 40% of patients.13 A favorable objective response to immunosuppression (eg corticosteroids) is seen in most patients.12
Individual case reports have recently described pituitary disease in patients with IgG4-RD.14–16 In a prospectively followed group of patients with IgG4-RD, we investigated the frequency and clinical presentation of cerebral disease.
Methods
All patients referred with a suspected diagnosis of IgG4-RD to the Gastroenterology Unit at University College Hospital, London, UK between March 2004 and October 2013 were followed prospectively. The diagnosis of IgG4-RD was made according to the Mayo Clinic HiSORt criteria.11 Neurological investigations were performed in patients with overt symptoms or clinical problems suggestive of cerebral disease. Other causes of cerebral disease were actively excluded. Imaging, blood tests and biopsies were performed according to clinical need. In patients with active disease, a reducing course of oral prednisolone was given, tailored to clinical response. Clinical relapse post-treatment was treated with the reintroduction of steroids and commencement of azathioprine (target dose 2 mg/kg/day). Clinical follow up was performed every 2–4 weeks during the initial steroid treatment phase, and subsequently every 3–6 weeks according to clinical progress.
Results
A diagnosis of IgG4-RD was made in 84 patients seen during the study period. All were treated with a course of steroids. Clinical cerebral disease was identified in three (4%) patients, details of which are reported below.
Case 1
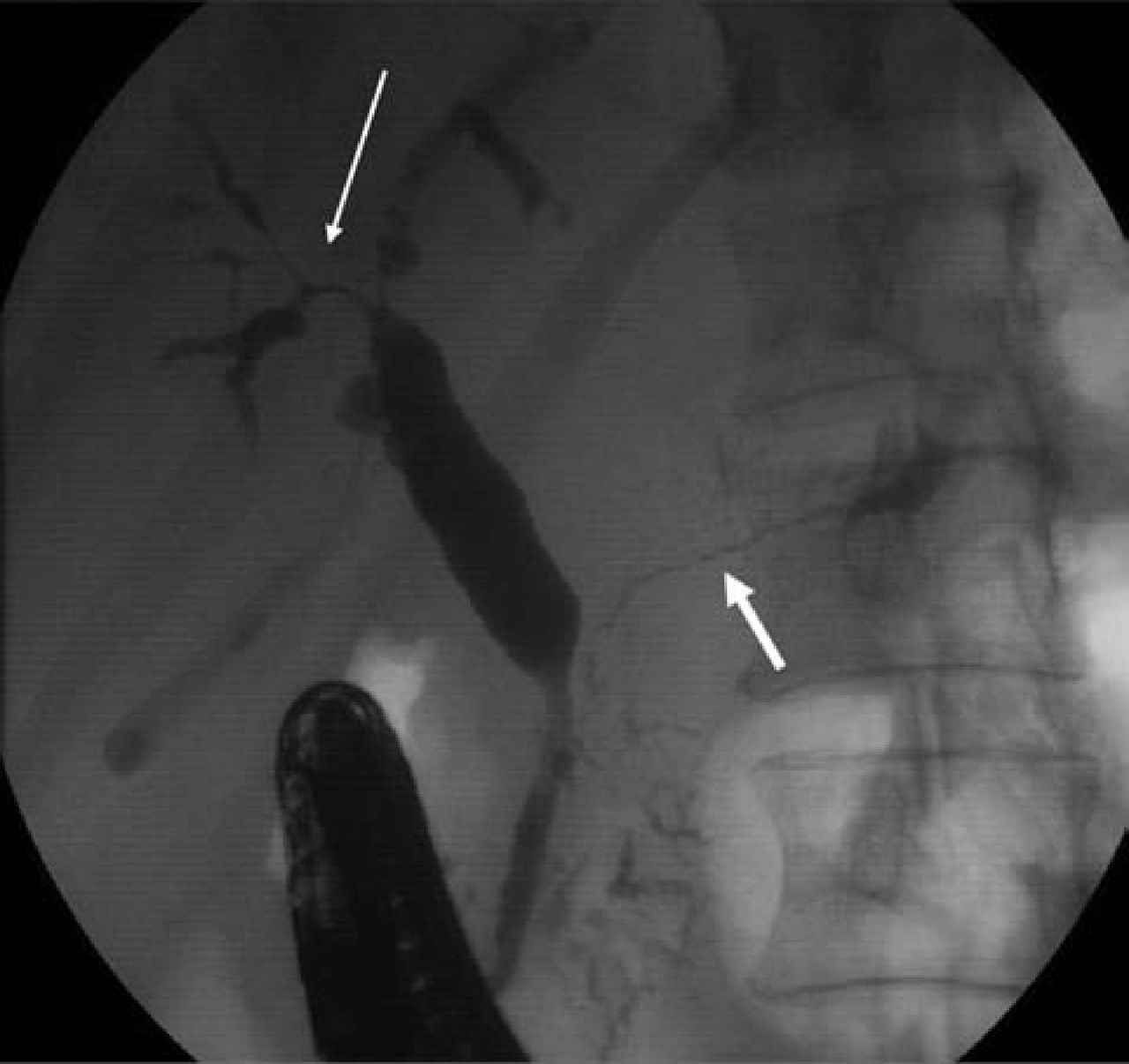
A 56-year-old male presented with malaise and abnormal liver function tests. Computed tomography (CT) imaging showed intrahepatic biliary dilatation, pancreatic enlargement, gastric and small bowel wall thickening, and mesenteric lymphadenopathy. Relevant blood tests showed a normochromic normocytic anaemia, cholestatic liver function, and a raised serum IgG4 of 2.3 g/l (normal range <1.3 g/l). Endoscopic retrograde cholangiopancreatography (ERCP) showed strictured and diffusely abnormal intrahepatic ducts and main pancreatic duct (Fig 1). Percutaneous biopsy of an enlarged submandibular salivary gland revealed a chronic lymphoplasmacytic infiltrate, with >10 IgG4-positive plasma cells per high-power film. The clinical picture was consistent with type-1 AIP and IgG4-RD, and the patient was commenced on a reducing course of prednisolone (30 mg daily). This led to biochemical, radiological and symptomatic improvement.
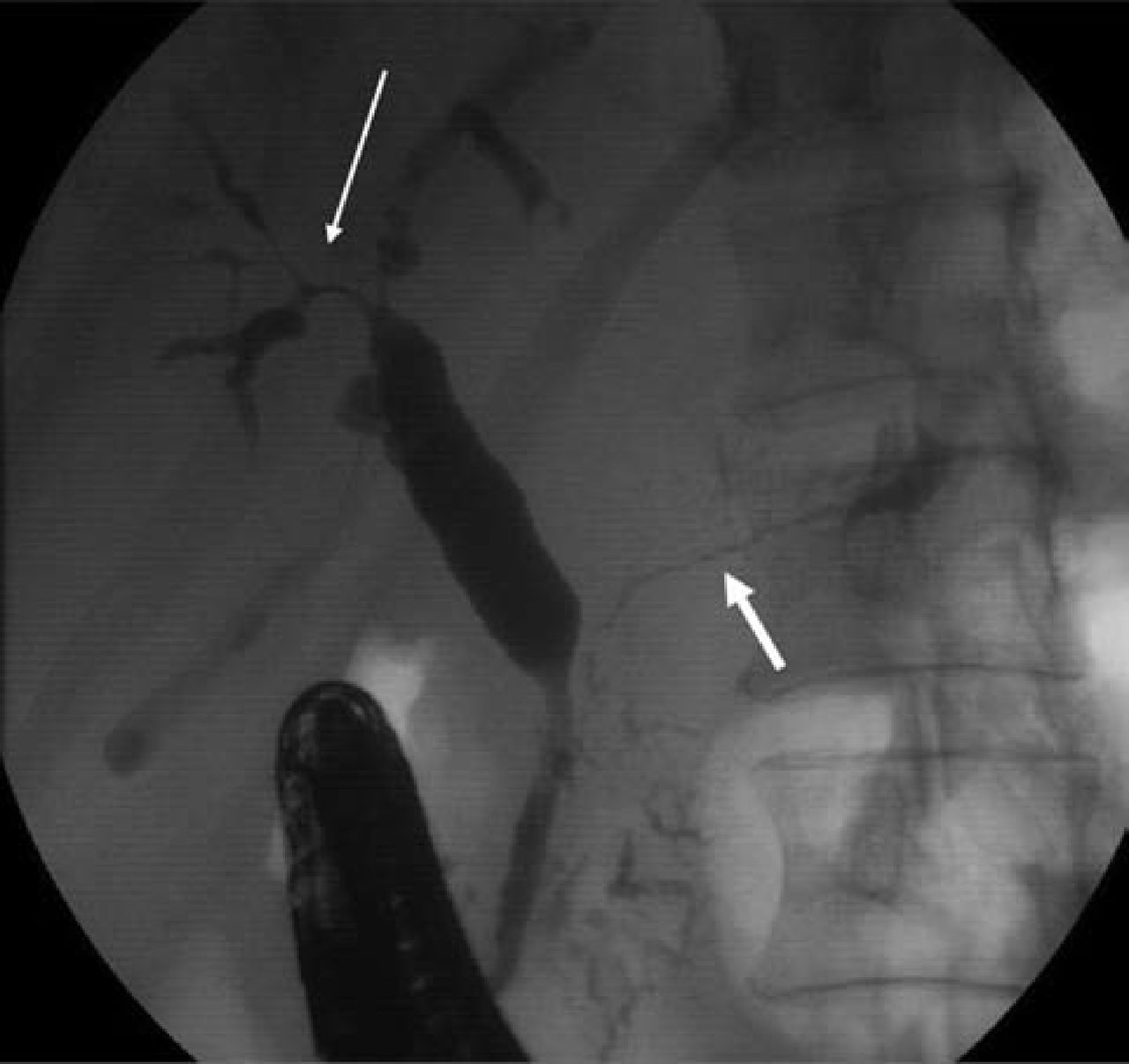
Endoscopic retrograde cholangiopancreatogram (case 1). Strictured and diffusely abnormal intrahepatic ducts (thin arrow) and main pancreatic duct (thick arrow) at the time of the initial presentation with jaundice.
During steroid reduction, the patient developed angina, with coronary angiography showing three-vessel coronary disease. He underwent coronary artery bypass surgery while on maintenance prednisolone 5 mg OD. Two weeks after an initially uneventful recovery, he developed confusion and a decreased consciousness level. Neurological and cardiac assessment, including a contrast CT scan, excluded a thromboembolic event. Serial electroencephalograms showed non-specific changes with generalised slow activity, indicating severe encephalopathy. Serial gadolinium-enhanced magnetic resonance imaging (MRI) suggested progressive diffuse periventricular and subcortical white matter signal change. The abnormal white matter showed small areas of restricted diffusion, and also areas of free diffusion with generalised volume loss, enlargement of ventricles and widening of sulci (Fig 2a). The cerebrospinal fluid (CSF) was clear, colourless, acellular, and had normal protein levels 0.32 g/l (normal range 0.15–0.45 g/l). No signs of systemic sepsis were evident. Antineuronal antibodies were negative. This neurological picture was thought to be most consistent with an ‘autoimmune encephalopathy’. Consent for a brain biopsy was not obtained.
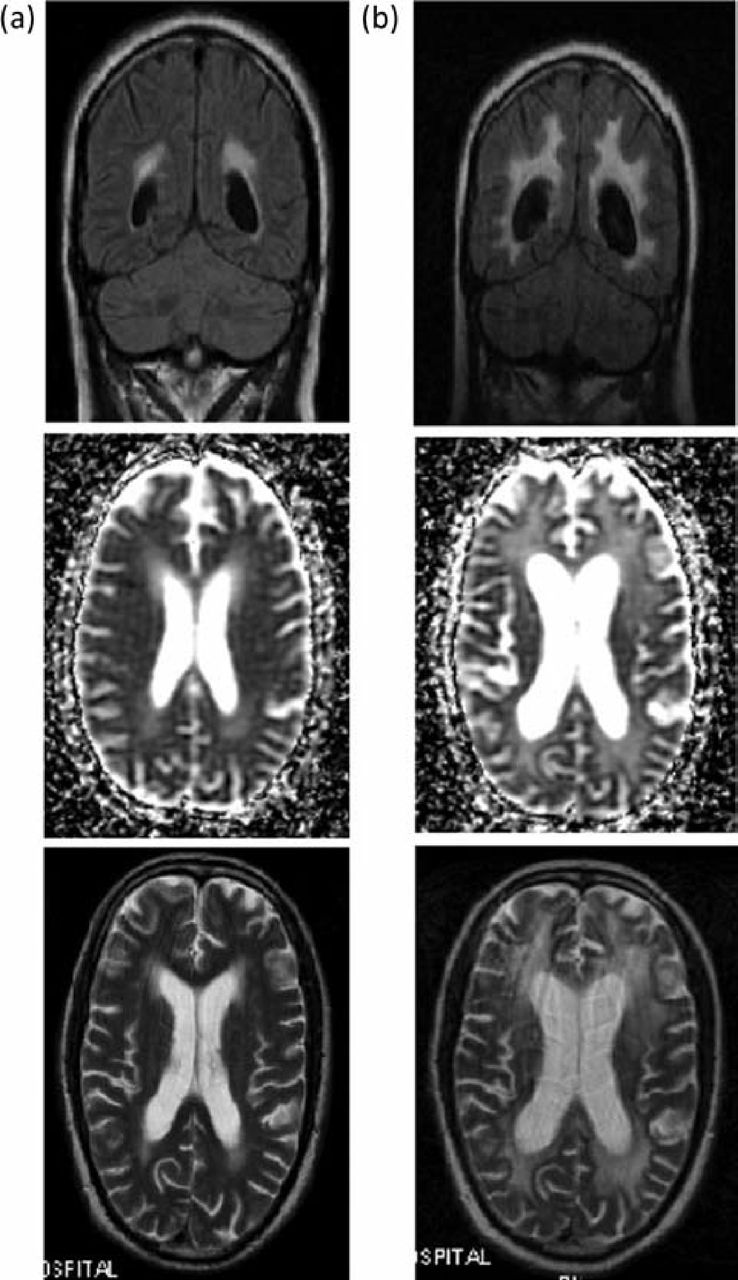
Gadolinium-enhanced MRI head (case 1). (a) Cerebral changes demonstrating increased abnormal white matter with small areas of restricted diffusion, and areas of free diffusion with generalised volume loss, enlargement of ventricles and widening of sulci. (b) Evolution of MRI changes was seen despite empirical treatment with high-dose steroid therapy, intravenous immunoglobulin, and antiviral and antifungal medication. MRI = magnetic resonance imaging.
Despite empirical treatment with high-dose steroid therapy, intravenous immunoglobulin, and antiviral and antifungal medication the patient showed no significant clinical improvement, although evolution of MRI changes was seen (Fig 2b). He remained dependant for all daily activities and was transferred to a long-term rehabilitation unit. The patient subsequently died, 11 months after the initial cerebral decompensation. A post-mortem was not performed.
Case 2
A 67-year-old man presented with obstructive jaundice. Imaging showed a low bile duct stricture that was associated with enlargement of the head of the pancreas. The clinical impression was that of carcinoma of the head of pancreas. At surgery, the head of pancreas could not be resected and therefore, a Roux-en-Y hepaticojejunostomy was performed. Intraoperative pancreatic biopsies showed chronic inflammation but no malignancy. Jaundice persisted post-operatively, with magnetic resonance cholangiopancreatography and percutaneous transhepatic cholangiography showing anastomotic and intrahepatic duct stricturing. The patient was subsequently referred to our unit, where serum IgG4 was shown to be elevated at 2.4 g/l. A diagnosis of type-1 AIP and IgG4-related cholangitis was made, and the patient's bilirubin normalised after commencing oral prednisolone 30 mg OD. Steroids were reduced and stopped after a five-month course.
Four months later he developed further jaundice. Percutaneous transhepatic cholangiogram showed worsening biliary strictures, but no improvement was seen with further percutaneous drainage and balloon dilatation. Liver biopsy showed large duct obstruction, severe fibrosis/pre-cirrhosis, and a lymphoplasmacytic infiltrate, with >10 IgG4-positive plasma cells per high-power film. Oral steroids were recommenced in addition to azathioprine (2 mg/kg/day) resulting in normalisation of liver function tests. Following slow reduction and cessation of steroids (over six months), and maintenance of azathioprine (at <1 mg/kg/day in view of nausea), the patient re-presented with a two-week history of profound weakness and malaise. There was no history of headache, fever, or jaundice. Examination revealed a heart rate of 80 beats/min and systemic blood pressure of 80/40 mmHg, but no other specific signs. The clinical impression was of adrenal insufficiency (which was assumed to be secondary to prolonged steroid use). Random cortisol was markedly low at <20 nmol/l (normal midnight level <140 nmol/l), but thyroid function tests showed low levels of T4 at 7.2 pmol/l (normal range 12–22 pmol/l) and thyroid stimulating hormone (TSH) at 0.7 mIU/l (range 0.27-4.2 mIU/l), suggesting possible pan-hypopituitarism. This diagnosis was confirmed, with results showing luteinizing hormone (LH) <0.1 IU/l (normal range 1.7–8.6 IU/l), follicle-stimulating hormone (FSH) 0.8 IU/l (normal range 1.5–12.4 IU/l), and testosterone <0.1 nmol/l (normal range 9.9–27.8 nmol/l).
Diabetes insipidus was also suggested by a history of polyuria and polydypsia, normal blood glucose, serum osmolality 294 mOsmol/kg (normal range 285–295 mOsmol/kg) and urine osmolality 263 mOsmol/kg (normal range 300–900 mOsmol/kg).
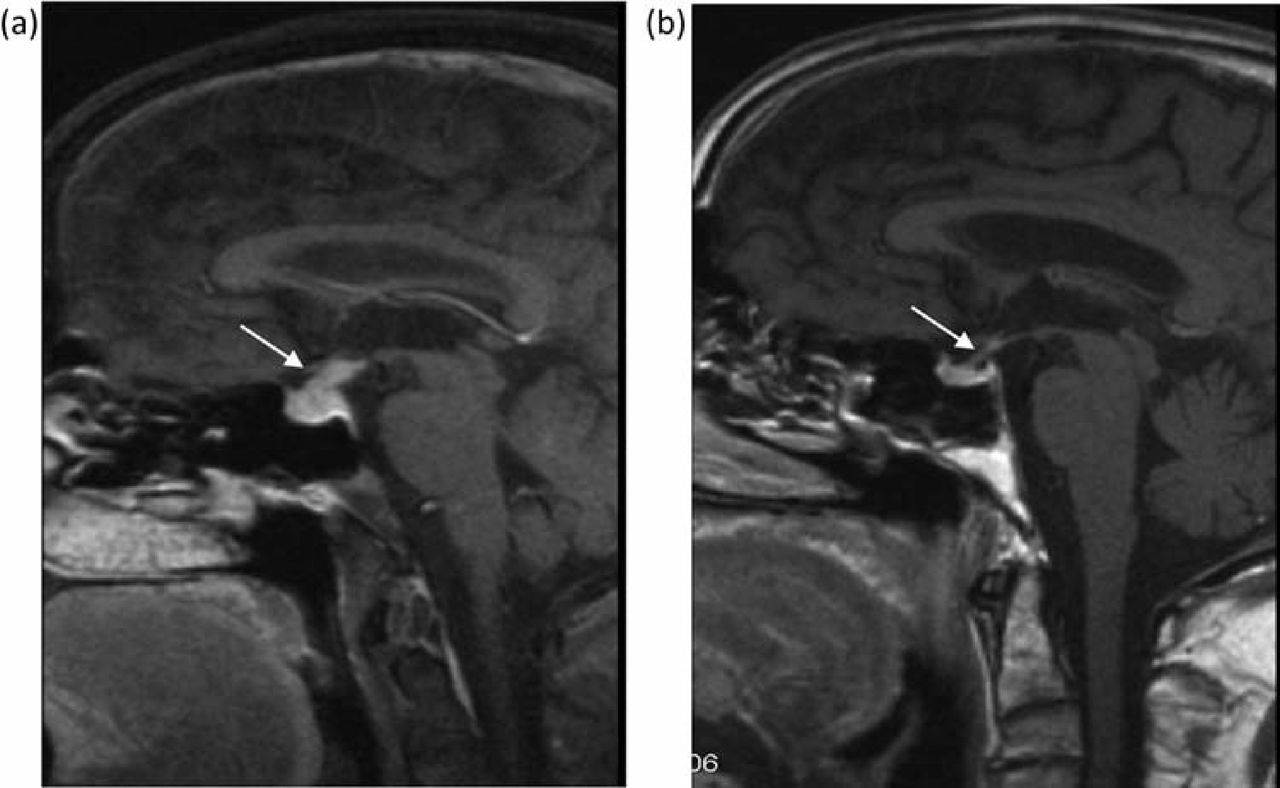
CT scan showed prominence of the pituitary stalk, but no other cerebral abnormality. Gadolinium-enhanced MRI showed enlargement and infiltration of the pituitary stalk, consistent with inflammation, rather than tumour (Fig 3a). The presumptive diagnosis of IgG4-related lymphoplasmacytic hypophysitis causing panhypopituitarism was made. The step-wise introduction of replacement hydrocortisone, fludrocortisone, testosterone, thyroxine and desmopressin led to resolution of symptoms. Prednisolone 30 mg was recommenced, as was azathioprine at 1.5 mg/kg/day, which led to complete resolution of the pituitary infiltrate on repeat gadolinium MRI six weeks later (Fig 3b). The patient remained clinically well on immunosuppression and had no further pituitary symptoms. Nevertheless, he represented eight months later with worsening jaundice in the absence of new dominant biliary strictures, and subsequently died from decompensated cirrhosis.
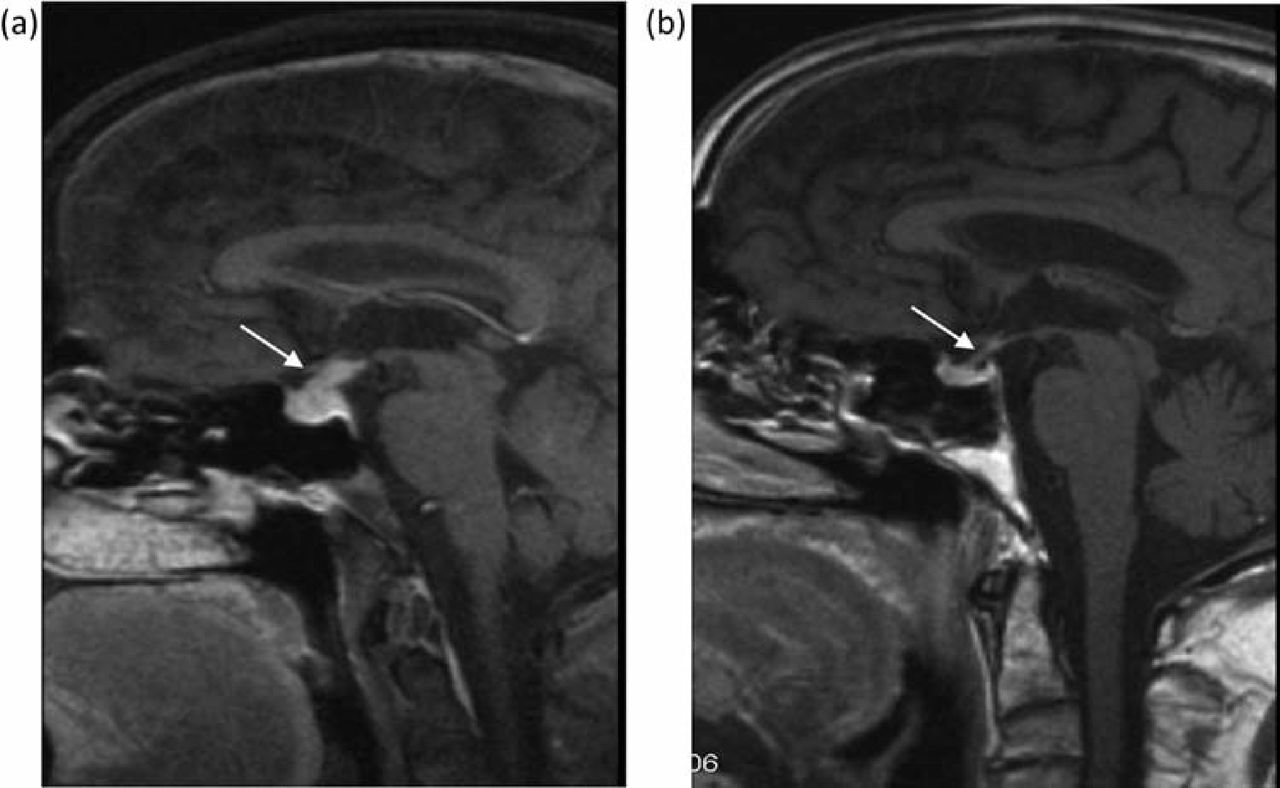
Gadolinium-enhanced MRI head (case 2). (a) Enlargement and infiltration of the pituitary stalk (arrow), consistent with inflammation. A presumptive diagnosis of IgG4- associated lymphoplasmacytic hypophysitis was made. (b) Resolution of the pituitary mass was seen six weeks after recommencing immunosuppression. MRI = magnetic resonance imaging.
Case 3
A 62-year-old Caucasian male presented to his local hospital with polydipsia and polyuria (but normal blood glucose levels), and evidence of hilar and mediastinal lymphadenopathy. A diagnosis of diabetes insipidus was made and the patient was commenced on desmopressin with symptomatic improvement. A CT of his abdomen demonstrated bilateral low-density renal lesions and abnormal tissue surrounding the abdominal aorta and iliac vessels, consistent with retroperitoneal fibrosis. A renal biopsy confirmed a diagnosis of tubulo-interstitial nephritis. Renal function remained normal (creatinine 119 μmol/l, estimated glomerular filtration rate (eGFR) >60 ml/min/1.73 m2). Subsequent immunostaining of the renal biopsy confirmed >10 IgG4-positive plasma cells per high-power film. Serum IgG4 levels were also raised (3.07 g/l). A pituitary MRI demonstrated a thickened pituitary stalk and a bulky anterior pituitary. LH, FSH and testosterone levels were normal. A diagnosis of IgG4-RD was made with renal (tubulo-interstitial nephritis), pituitary (hypophysitis) and retroperitoneal (retroperitoneal fibrosis) involvement. The patient was referred to our service at University College London Hospital and was commenced upon prednisone 40 mg OD. A deterioration in renal function was noted at this time point (creatinine 543 μmol/l, eGFR 9 ml/min/1.73 m2) but this function subsequently improved following the introduction of steroids. The patient remains well on a low dose of prednisone 10 mg OD with stable renal function (creatinine 148 μmol/l, eGFR 44 ml/min/1.73 m2) but remains reluctant to commence azathioprine.
Discussion
Over the past ten years, it has become accepted that AIP is a global disease,11,17,18 and that it is one component of a multi-organ IgG4-RD, in which an IgG4-positive lymphoplasmacytic infiltrate within affected tissues is a characteristic feature.11,19,20 Cerebral involvement in IgG4-RD is rarely reported, with most cases relating to either retro-orbital involvement (with associated infra-orbital nerve swelling21 and visual field defects22) or to pituitary disease (Table 1).15,16,23 Non-infectious hypertrophic pachymeningitis, inflammation and subsequent thickening of the dura mater (cranial or spinal) is regarded as another neurological manifestation of IgG4-RD.24,25
Clinical presentation of the cerebral manifestations of IgG4-related disease.
In our series, we found neurological involvement in 4% of patients. Clearly this may not represent the true prevalence of neurological disease in IgG4-RD as those patients who are referred to a tertiary referral centre may form a skewed sub-sample of the patient population. Despite this caveat, the brain appears to be a much less common site of involvement than other sites, including the biliary tree, kidneys, salivary glands and retroperitoneum.26,27 In the cases reported here, unequivocal evidence of an IgG4 inflammatory infiltrate causing neurological disease could not be obtained because of a lack of cerebral histology. Nevertheless, other causes were intensively sought and excluded, and imaging strongly suggested an inflammatory cause. Case reports have recently suggested pituitary involvement in AIP/IgG4-RD diagnosed histologically following pituitary resection.15 In case 2, it may be that treatment with steroids delayed the clinical presentation of pituitary failure, which occurred only after steroid withdrawal. Swelling of the pituitary stalk may also be seen in sarcoidosis, tuberculosis, Wegner's granulomatosis and lymphocytic hypophysitis.
In cases 1 and 2, neither patient showed clinical evidence of worsening pancreatic disease at the time of active neurological disease. This supports the observation that clinical relapse in IgG4-RD most commonly occurs in extrapancreatic sites.28,29 The findings also suggest that disease control in one organ (with or without treatment) cannot be assumed to be paralleled in other tissues. Our observation in these two cases was mirrored by another case report in which symptomatic pituitary failure related to lymphocytic hypohysitis developed despite maintenance steroid therapy for AIP.16 Case 3 presented primarily with symptoms of diabetes insipidus and on subsequent imaging was found to have renal abnormalities and evidence of retroperitoneal fibrosis.
Disease relapse is now recognised to occur in at least 40% of AIP/IgG4-RD patients,28,30,31 but appropriate management of this is yet to be defined. It is of note that our patient with panhypopituitarism was on maintenance azathioprine at presentation (albeit at a low dose). The fact the patients in both cases 1 and 2 subsequently died of extra-pancreatic complications supports the argument that those with significant multi-organ involvement may have a more progressive disease. These patients may be more difficult to manage with a single course of steroids than those with disease confined to the pancreas.28,31,32
We believe that this report emphasises the need to maintain a high-level clinical vigilance for and awareness of the myriad of clinical manifestations of IgG4-RD, including neurological disease. These may occur at presentation or upon follow up.
- © 2015 Royal College of Physicians
References








{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.