
ABSTRACT
Isolated microscopic, or non-visible, haematuria of glomerular origin was previously regarded a benign finding, but it is now known that, even in the absence of proteinuria, hypertension or renal impairment at presentation, haematuria is associated with increased risk of kidney failure in the long term. The most common causes of isolated microscopic haematuria among children and young adults are IgA nephropathy, Alport syndrome (AS), and thin basement membrane nephropathy (TBMN). AS, which is usually inherited as an X-linked or autosomal recessive trait, and TBMN, which is usually autosomal dominant, are caused by mutations in the genes encoding type-IV collagen, an abundant component of the glomerular basement membrane. A detailed family history with screening of at-risk relatives is important, allowing prompt diagnosis of affected relatives and helping determine the mode of transmission. As costs fall and availability increases, genetic testing is increasingly being used in clinical practice to provide diagnostic and predictive information for patients and their families.
Isolated microscopic haematuria should not be considered a ‘benign’ finding as it is associated with an increased risk of chronic kidney disease in later life.
The most common causes of isolated microscopic haematuria in children and young adults are IgA nephropathy, thin basement membrane nephropathy and Alport syndrome.
Urinary dipstick screening should be offered to family members where an inherited cause is known or suspected, both to assess their risk of kidney disease and also allow mode of transmission to be determined.
The combination of detailed family history, kidney biopsy or genetic testing can allow a diagnosis to be made in a large proportion of patients, and this can inform prognosis in an individual and risk to family members or future children.
Individuals with persistent microscopic haematuria, whatever the cause, should have long-term follow up to monitor renal function and control blood pressure and proteinuria.
Introduction
Microscopic or non-visible haematuria is defined as two or more red cells per high-power field present in a mid-stream urine sample on more than one occasion, and unrelated to exercise, trauma or menstruation. Prevalence ranges between 0.18 and 16.1% depending on the population.1 Haematuria can result from bleeding anywhere from the glomerulus to urethra, and can be divided into glomerular or non-glomerular causes. Non-glomerular causes include malignancy, stones, papillary necrosis and metabolic disorders, most of which present with loin pain or abnormalities on imaging. Older adults (especially smokers) should initially be investigated with renal imaging and cystoscopy to exclude urothelial or renal malignancy, whereas non-neoplastic kidney disease is more likely in those under 40. Guidelines for referral to urology or nephrology in those with microscopic haematuria are listed in Box 1.
Referral guidelines for patients with haematuria. Adapted with permission.2
Microscopic haematuria of glomerular origin can occur with proteinuria, hypertension or renal dysfunction, which indicate kidney damage. If these are present, referral to nephrology for investigation (usually with kidney biopsy) is indicated because this allows diagnosis of disorders such as glomerulonephritis, which can be renal-limited or part of a systemic disorder, and which may require specific therapy (eg immunosuppression).
Isolated microscopic haematuria refers to haematuria in the absence of proteinuria, hypertension or renal dysfunction, and in this situation a decision to pursue invasive investigation is less straightforward. Historically, isolated microscopic haematuria was regarded as ‘benign’ but, while the short-term risk of kidney failure is undoubtedly small, it is now known that the lifetime risk is significantly higher than that of the general population: in one large study, isolated microscopic haematuria in healthy young adults was associated with a substantially increased risk (adjusted hazard ratio 18.5 over 22 years) of developing end-stage renal disease (ESRD) although this was small in absolute terms (0.7% in those with asymptomatic microscopic haematuria compared to 0.04% without).3
The commonest cause of isolated microscopic haematuria of glomerular origin is probably IgA nephropathy (IgAN); however a number of genetic disorders can also present similarly, including Alport syndrome (AS) and thin basement membrane nephropathy (TBMN). Accurate diagnosis at presentation can inform prognosis, guide appropriate treatment or long-term follow up as well as allow appropriate counselling and screening of at-risk family members. With falling genetic sequencing costs, and increasing understanding of the genes responsible for disease, the role of genetic testing in clinical practice is growing.
IgA nephropathy
IgAN is the most common glomerulonephritis worldwide, with an incidence of 10–20 per million population in the UK4 and more in some Asian populations.5 IgAN is not usually inherited, but approximately 5% of patients have a relative with evidence of kidney disease. IgAN characteristically presents with microscopic haematuria, sometimes with synpharyngitic macroscopic haematuria (visible haematuria coinciding with respiratory tract infections), proteinuria or renal impairment. Diagnosis requires a kidney biopsy demonstrating IgA-dominant or co-dominant immune deposits with evidence of accompanying inflammation in the glomeruli. Approximately 20% of patients with IgAN develop ESRD within 20 years of diagnosis.6 Although the prognosis is substantially better in those with IgAN and isolated microscopic haematuria,7 it remains an independent risk factor for ESRD8 and follow-up for at least 10 years is recommended.9 Guidelines advise supportive management with renin–angiotensin system (RAS) blockade, followed by a trial of corticosteroids if there is persistent proteinuria with preserved renal function,10 although preliminary results from a large, prospective randomised controlled trial do not support steroid use.11
Type-IV collagen nephropathies
Type-IV collagen is a major component of basement membranes, consisting of a macromolecular network of triple helices formed from combinations of alpha chains, each encoded by a separate gene. The α3.α4.α5 type-IV collagen heterotrimer is encoded by the genes COL4A3, COL4A4 (chromosome 2) and COL4A5 (X chromosome) and is the most abundant constituent of the glomerular basement membrane (GBM). Mutations of these genes are the commonest cause of familial microscopic haematuria.
Alport syndrome
AS has a prevalence of 1 in 50,000 live births and characteristically presents with persistent microscopic haematuria in childhood, often with episodes of macroscopic haematuria. 70% of patients develop sensorineural hearing loss that mirrors renal impairment in severity, as well as ocular abnormalities including anterior lenticonus (30%) and retinal flecks (50%). Although significant visual impairment is not a feature, deafness can be a significant problem and its early identification in children is important because appropriate support can prevent educational delay.
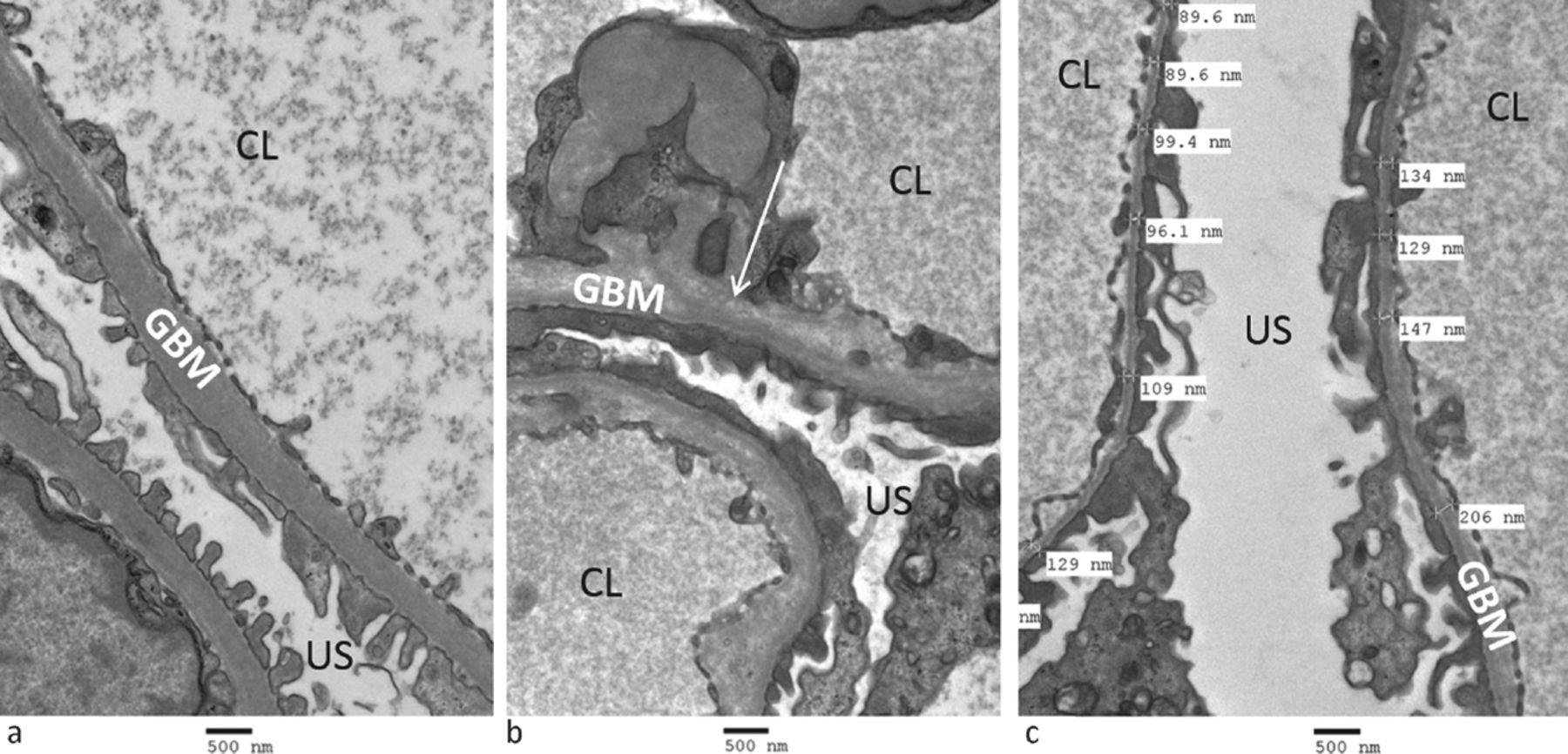
Diagnosis of AS can be made by kidney biopsy, with electron microscopy demonstrating areas of irregular GBM thickness with lamellation, splitting and sometimes characteristic ‘basket-weave’ appearances (Fig 1). Focal segmental glomerulosclerosis may also be seen on light microscopy in the latter stages of disease. 85% of AS is caused by mutations of COL4A5 and therefore exhibits X-linked inheritance. Penetrance is very high in males, with the vast majority reaching ESRD before age 40.12 95% of female carriers of X-linked AS have haematuria, and although their prognosis is better, up to 15% develop ESRD by age 60.13 15% of AS is autosomal recessive, caused by homozygous or compound heterozygous mutations of the autosomal COL4A3 or COL4A4 genes. Carriers of a heterozygous autosomal COL4A3/COL4A4 mutation and women harbouring a heterozygous X-linked COL4A5 mutation have a similar lifetime risk of kidney failure (10–15%);13 however the risk to their male offspring is clearly very different. Autosomal dominant AS has been reported, associated with heterozygous COL4A3 or COL4A4 mutations, but is clinically difficult to distinguish either from the carrier state of autosomal recessive AS or from autosomal dominant TBMN (see below).14 In addition, digenic inheritance, where mutations at two loci on two different genes interact to produce disease, has recently been proposed to explain the variable genotype–phenotype correlation and incomplete penetrance often seen in AS.15

Electron microscopy of a kidney biopsy. a) normal glomerular basement membranes; b) a patient with Alport syndrome, showing disruption of the glomerular basement membrane architecture with lucent inclusions (arrow); and c) a patient with thin basement membrane nephropathy showing marked thinning of the glomerular basement membrane (thickness <200 nm is abnormal). CL = capillary lumen; GBM = glomerular basement membrane; US = urinary space.
AS can be confirmed by genetic testing and the introduction of next-generation sequencing panels enables simultaneous, cost-efficient sequencing of COL4A3, COL4A4 and COL4A5. This is especially valuable where the inheritance pattern in a family does not distinguish X-linked from recessive disease, since the risk to future (male) children is much greater in the former.
Management of AS includes blood pressure control, RAS blockade for patients with persistent proteinuria or microalbuminuria, and avoidance of nephrotoxic or ototoxic medications.13,16,17 Family members should be screened for microscopic haematuria and, where appropriate, offered genetic testing to assess risk of kidney disease to them and their future offspring.15
Thin basement membrane nephropathy
TBMN affects 1% of the population and is characterised by persistent isolated haematuria with thinned GBMs on kidney biopsy (Fig 1). Affected individuals frequently have a family history of haematuria or kidney disease that is consistent with autosomal dominant inheritance, and approximately 40% harbour a heterozygous mutation of COL4A3 or COL4A4.18
Although the renal outcome is generally good, recent studies show that carriers of heterozygous COL4A3/COL4A4 mutations are at increased risk of ESRD (up to 14% by age 60).13,19,20 Where proteinuria and/or renal impairment is present, kidney biopsy may show focal segmental glomerulosclerosis on light microscopy. It is unclear why some individuals with TBMN develop renal impairment while most do not.
Autosomal recessive AS has been reported in offspring of parents who both have autosomal dominant TBMN,20,21 and while it has not been established whether all heterozygous carriers of AS mutations have TBMN, and vice versa, there is clearly substantial overlap between these categories. For this reason, screening of family members (including spouses) is essential in patients with known or suspected TBMN – both to identify early evidence of renal disease in at-risk relatives and also to clarify mode of transmission: observing male-to-male transmission excludes X-linked AS, which in women can be indistinguishable from TBMN.
The reported benefits of RAS blockade in heterozygous carriers of autosomal or X-linked Alport mutations suggests treatment with these agents in patients with TBMN and hypertension, proteinuria or renal impairment is beneficial.13 Given their increased lifetime risk of kidney failure, patients with TBMN should be followed up long term. Although not absolutely contraindicated, living kidney donation by patients with TBMN or carriers of AS is generally avoided.
Other rare causes of familial haematuria
Additional rare autosomal dominant diseases presenting with microscopic haematuria and associated with ESRD are known, highlighting the importance of accurate diagnosis in patients who may previously have been labelled as having ‘benign familial haematuria’.
Hereditary angiopathy, nephropathy, aneurysms and muscle cramps (HANAC) syndrome is an extremely rare autosomal dominant condition caused by heterozygous mutations of the COL4A1 gene resulting in nephropathy characterised by kidney cysts, haematuria and a low risk of ESRD.22 Additional clinical features include cerebral angiopathy and aneurysms, muscle cramps with elevated serum creatine kinase and retinal haemorrhages.
MYH9 (nonmuscle myosin heavy chain IIA)-associated disease underlies Fechtner and Epstein syndromes, characterised by macrothrombocytopaenia (the May–Hegglin platelet anomaly), glomerulopathy and sensorineural deafness. Prevalence is less than 1 in 100,000 and renal involvement manifests with microscopic haematuria and/or proteinuria and renal dysfunction. 30% of patients develop ESRD, usually as young adults.23
CFHR5 nephropathy is an autosomal dominant disorder common in Greek Cypriots; it is extremely rare outside this community. It presents with microscopic haematuria with minimal or no proteinuria and episodes of synpharyngitic macroscopic haematuria (reminiscent of IgAN). Kidney biopsy demonstrates C3 glomerulonephritis (with deposition of complement but not immunoglobulins in glomeruli). It is caused by mutations of the CFHR5 gene which encodes a protein important in the regulation of complement, part of the innate immune system.24 Approximately 80% of affected men will develop ESRD, usually after age 40. For unknown reasons, fewer than 20% of affected women develop ESRD.25
Conclusion
Microscopic haematuria is a relatively common finding and, even in the absence of proteinuria, hypertension or renal impairment, is now known to be associated with significantly increased risk of kidney failure, with the risk depending on the underlying cause. A detailed family history is vital in assessing patients and urine dipstick screening of at-risk family members can help establish the diagnosis (Fig 2). Genetic testing is a useful diagnostic tool that allows cascade screening of relatives, predicts risk to future children, and can reduce the need for invasive kidney biopsies.

Assessment and diagnosis of a patient with persistent haematuria, where infection and structural renal tract disease (eg malignancy) has been excluded. Kidney biopsy is not usually indicated in the absence of proteinuria, hypertension or renal impairment, but may be performed in some circumstances. AS = Alport syndrome; TBMN = thin basement membrane nephropathy.
Acknowledgements
DPG is supported by the Medical Research Council. The authors would like to thank Innes Clatworthy for assistance with electron microscopy.
- © Royal College of Physicians 2015. All rights reserved.








{kind=link}
{kind=link}