
ABSTRACT
There is a growing understanding of the epigenetic mechanisms that regulate gene expression in healthy conditions and a realisation that dysregulation of these mechanisms is an underlying factor in many human diseases. We discuss studies demonstrating that small molecule inhibitors of epigenetic regulatory proteins can block pathogenic mechanisms associated with rheumatoid arthritis, focusing on the effects of these inhibitors on synovial fibroblasts – fibroblast-like synoviocytes.
Rheumatoid arthritis and the role of synovial fibroblasts
Rheumatoid arthritis is a systemic and chronic inflammatory disease characterised by pain, swelling, and destruction of synovial joints. Inflammation of the synovial membrane (synovitis) is associated with infiltration by leukocytes of multiple lineages, including T cells, B cells, macrophages and dendritic cells.1 These immune cells can produce a range of mediators that contribute to synovitis, including multiple pro-inflammatory cytokines secreted by T cells and macrophages, and auto-antibodies produced by plasma cells. In addition to leukocyte infiltration, the intimal lining layer, which in health is 2–3 cells thick, undergoes marked hyperplasia in rheumatoid arthritis, associated with a dramatic expansion of synovial fibroblasts or fibroblast-like synoviocytes (FLS). The properties of these fibroblasts are substantially different from those in a healthy synovium, with the cells demonstrating a loss of contact inhibition and anchorage-dependent growth, reduced apoptosis and increased proliferation.2,3
Fibroblasts are thought to be key mediators of tissue damage in rheumatoid arthritis through their secretion of matrix metalloproteinases and cathepsins that degrade cartilage and bone.4,5 Additionally, rheumatoid arthritis fibroblasts produce factors that enhance the differentiation of bone-absorbing osteoclasts and inhibit the function of bone-forming osteoblasts, thus promoting bone erosion. With time, fibroblast hyperplasia can cause the intimal lining layer to develop into a mass of pannus tissue rich in fibroblasts and osteoclasts that invades the adjacent articular cartilage and subchondral bone. These processes are accompanied by neovascularisation of the synovial sublining, which facilitates leukocyte infiltration.
Bidirectional signalling between immune cells and synovial fibroblasts seems to have a key role in the perpetuation of joint inflammation. Thus, although inflammatory mediators released by infiltrating leukocytes promote fibroblast hyperplasia, synovial fibroblasts express factors that maintain the recruitment and survival of immune cells within the inflamed joint.6 Notably, however, there is considerable heterogeneity in the cellular composition of synovial tissue in patients with rheumatoid arthritis. In some cases, few infiltrating leukocytes are noted despite fibroblast hyperplasia, swollen and tender joints and the presence of auto-antibodies.
Evidence for a clear link between the invasive properties of synovial fibroblasts and joint destruction was demonstrated in a study showing the ability of FLS harvested from the synovial tissue of patients with rheumatoid arthritis to invade a Matrigel basement membrane matrix (as a mimic of cartilage) in vitro.7 By obtaining samples at multiple times and from different joints, the authors showed that measured invasiveness varied much less between FLS obtained from the same patients than between those from different patients. Hence, the in vitro invasiveness of FLS is an intrinsic patient characteristic. Most importantly, when comparing patients with the most or least invasive FLS, the authors showed a clear and statistically significant association between invasiveness of the FLS and the radiologically estimated yearly rate of joint destruction. Thus, important FLS characteristics, such as invasiveness, can vary between patients. This variation has important implications for disease progression.
Heterogeneity among rheumatoid arthritis FLS at the molecular level has been shown in a study8 in which gene expression patterns in cultured FLS derived from the synovia of 19 different patients were analysed. Using complementary DNA microarrays to measure the expression of approximately 24,000 genes, the authors classified FLS into two major groups on the basis of differential gene expression. Prominent gene functions that were overrepresented among group I FLS were those associated with autocrine signalling in cell proliferation and survival, the alternative pathway of complement activation, oxidative stress and oncogenesis. Conversely, group II FLS were characterised by increased expression of several genes linked to the transforming growth factor β pathway, a gene signature that is a feature of a subtype of fibroblasts called myofibroblasts. In accordance with this finding, group II FLS express α-smooth muscle actin, a property of myofibroblasts but not fibroblasts.
The two groups of FLS were associated with distinct characteristics in synovial tissues. Thus, group I FLS arose in tissues with a low degree of inflammation, whereas group II FLS were found in highly inflamed synovia. The implication of these observations is that the FLS gene signatures and related functional properties are directed by the local tissue microenvironment. Such environmental control is mediated by epigenetic mechanisms, which alter gene expression patterns and cellular function without changing the DNA sequence. Before we discuss how these mechanisms can be targeted to treat rheumatoid arthritis, we briefly describe some basic aspects of epigenetics.
Epigenetic mechanisms
Three main classes of epigenetic mechanisms have been described. Firstly, various non-protein-coding RNA species (ncRNA), including micro RNA (miRNA) and long non-coding RNA (lncRNA), can regulate expression of protein-coding genes at both transcriptional and post-transcriptional levels. Because individual ncRNAs typically control many genes, changes in expression of ncRNA can have a major impact on the overall pattern of gene expression within a cell. Secondly, nucleotides within DNA can undergo modifications, the best characterised of which is the methylation of cytosine residues. Cytosine methylation occurs predominantly in the context of the dinucleotide sequence CpG, which is frequently enriched in regulatory regions of DNA, such as gene promoter regions. In such a setting, DNA methylation typically leads to suppression of gene transcription. Finally, histone proteins with which the DNA is closely associated in chromatin are subject to a wide variety of post-translational modifications. As discussed further below, histone modifications alter the accessibility of DNA to the transcriptional machinery – and hence gene expression – through a range of mechanisms. Although miRNA expression, DNA methylation and histone modifications are all altered in FLS in rheumatoid arthritis,9–12 we focus on the relevance of histone modifications.
Many different amino acids within histones can be modified after translation. Most modifications occur in the N-terminal or C-terminal tails. These tails extend outside the main, globular histone domains, around which DNA is wrapped. As many as 60 (by some accounts) different chemical modifications have been documented. The most common are acetylation, methylation, phosphorylation, ubiquitination, sumoylation, adenosine diphosphate ribosylation, crotonylation and biotinylation.13
Three classes of proteins – commonly referred to as ‘writers’, ‘erasers’ and ‘readers’ – have roles in mediation of epigenetic regulation through the post-translational modification of histones. Epigenetic writers are enzymes that add specific modifications to amino acids within histones, whereas erasers are enzymes that remove these modifications. Epigenetic readers are proteins that can recognise and bind specifically to histone proteins on the basis of the post-translational modifications present. Large families of writers, erasers and readers have been identified that add, remove and recognise specific modifications in the context of particular amino acid sequences.
Because of the capacity for many histone residues to be modified and the diversity of possible modifications, the decoration of histones has been likened to an epigenetic code, which can be deciphered by analysing the binding of reader proteins. Reader proteins translate this code into gene expression patterns by bringing with them activities that facilitate or hinder gene expression – for instance, enzymatic activities that remove or add histones to specific sites on chromatin, making those regions of DNA more or less accessible to transcription factors. These chromatin-modifying activities can be intrinsic to the reader proteins or might be present in other proteins that are recruited with the readers as part of larger multiprotein complexes. Through mechanisms such as these, the genome-wide activity of writers, erasers and readers controls cell-type-specific gene expression profiles, and alterations in epigenetic regulatory mechanisms can lead to aberrant gene expression and disease.
Coincident with the growing understanding of the role of epigenetics in the regulation of gene expression and cell function in health and disease has been the discovery that small molecule inhibitors targeting histone writers, erasers and readers can be developed. Here we describe our studies of inhibitors of erasers and readers to investigate the potential to suppress the pathogenetic behaviour of FLS in rheumatoid arthritis.
Inhibiting erasers: effects of histone deacetylase inhibition on FLS function
Eighteen enzymes have been identified that are capable of deactylating acetylated lysine residues, including those found within histones. Small molecules that inhibit histone deacetylases have been developed, and most are active against several histone deacetylases. Several of these molecules have shown effects when tested in rheumatoid arthritis synovial samples in vitro, suggesting therapeutic potential. These effects include suppression of the pro-inflammatory cytokines interleukin (IL)-6 and tumour necrosis factor by rheumatoid arthritis synovial macrophages and synovial tissue explants,14 inhibition of FLS proliferation15 and sensitisation of FLS to induction of cell death through proapoptotic stimuli.16
We investigated the effects of histone deacetylase inhibitors on expression of IL-6 by rheumatoid arthritis FLS stimulated with either pro-inflammatory cytokines (IL-1β, tumour necrosis factor) or toll-like receptor ligands (lipopolysaccharide, polyinosinic:polycytidylic acid) as mimics of infection.17 Two different histone deacetylase inhibitors, trichostatin A and givinostat (ITF2357), strongly inhibited the secretion of IL-6 by rheumatoid arthritis FLS induced by treatment with either pro-inflammatory cytokines or microbial stimuli (Fig 1). Cell viability was not affected by treatment, suggesting that inhibition of cytokine production was not an indirect effect of inducing cell death. Inhibition of IL-6 expression was observed at the messenger RNA (mRNA) level: histone-deacetylase-inhibitor-mediated reduction of IL-6 mRNA was apparent at 4 hours and, more prominently, 8 hours post-stimulation (Fig 2A). These results suggested that the histone deacetylase inhibitors might be inhibiting IL-6 gene transcription. However, by analysing time-dependent mRNA decay in IL-6, it was found that the degradation of mRNA was significantly accelerated in trichostatin-A-treated FLS compared with control cells (Fig 2B–D). Thus, inhibition of IL-6 expression in FLS by histone deacetylase inhibitors is mediated at least partly by modulation of mRNA stability. Histone deacetylases associate in protein complexes with factors that regulate mRNA stability,18 but the precise mechanisms by which inhibitors affect the stability of IL-6 mRNA remain to be determined.
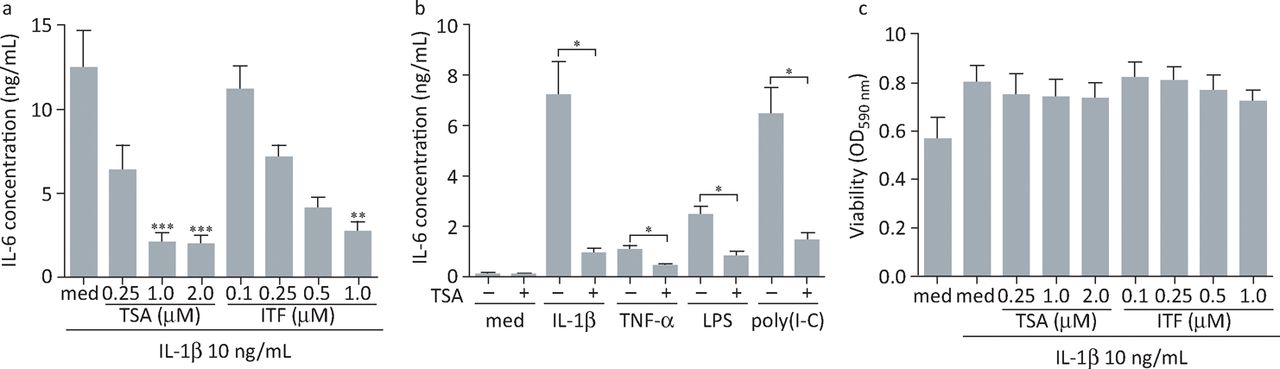
Histone deacetylase inhibitors suppress IL-6 production by rheumatoid arthritis FLS without affecting cell viability. (A) FLS were stimulated with 10 ng/mL of IL-1β alone or in the presence of increasing concentrations of TSA or ITF2357 for 24 hours (n = 6–10). (B) Cells were left untreated or pre-exposed to 1 μM TSA followed by 24 hours of stimulation with IL-1β (10 ng/mL), TNFα (10 ng/mL), LPS (1 μg/mL) or poly(I:C) (10 μg/mL) (n = 4). IL-6 concentrations in cell-free tissue culture supernatants were determined by enzyme-linked immunosorbent assay. Results, presented as mean (± standard error) IL-6 concentration, were either subjected to the Kruskal–Wallis test followed by Dunns’ multiple comparison analysis, with cells not treated with histone deacetylase inhibitors used as reference controls (A), or to the Mann-Whitney U test (B). *p<0.05; **p<0.01; ***p<0.001. (C) Viability of cells after 24 hours of stimulation with IL-1β (10 ng/mL) in the presence or absence of increasing doses of TSA or ITF2357 was assessed by measurement of MTT reduction (n = 5). Values representing changes in MTT processing are shown as mean (± standard error) optical density at 590 nm. FLS = fibroblast-like synoviocytes; IL = interleukin; LPS = lipopolysaccharide; MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; OD = optical density; Poly(I:C) = polyinosinic:polycytidylic acid; TNFα = tumour necrosis factor α; TSA = trichostatin A. Reproduced with permission.17
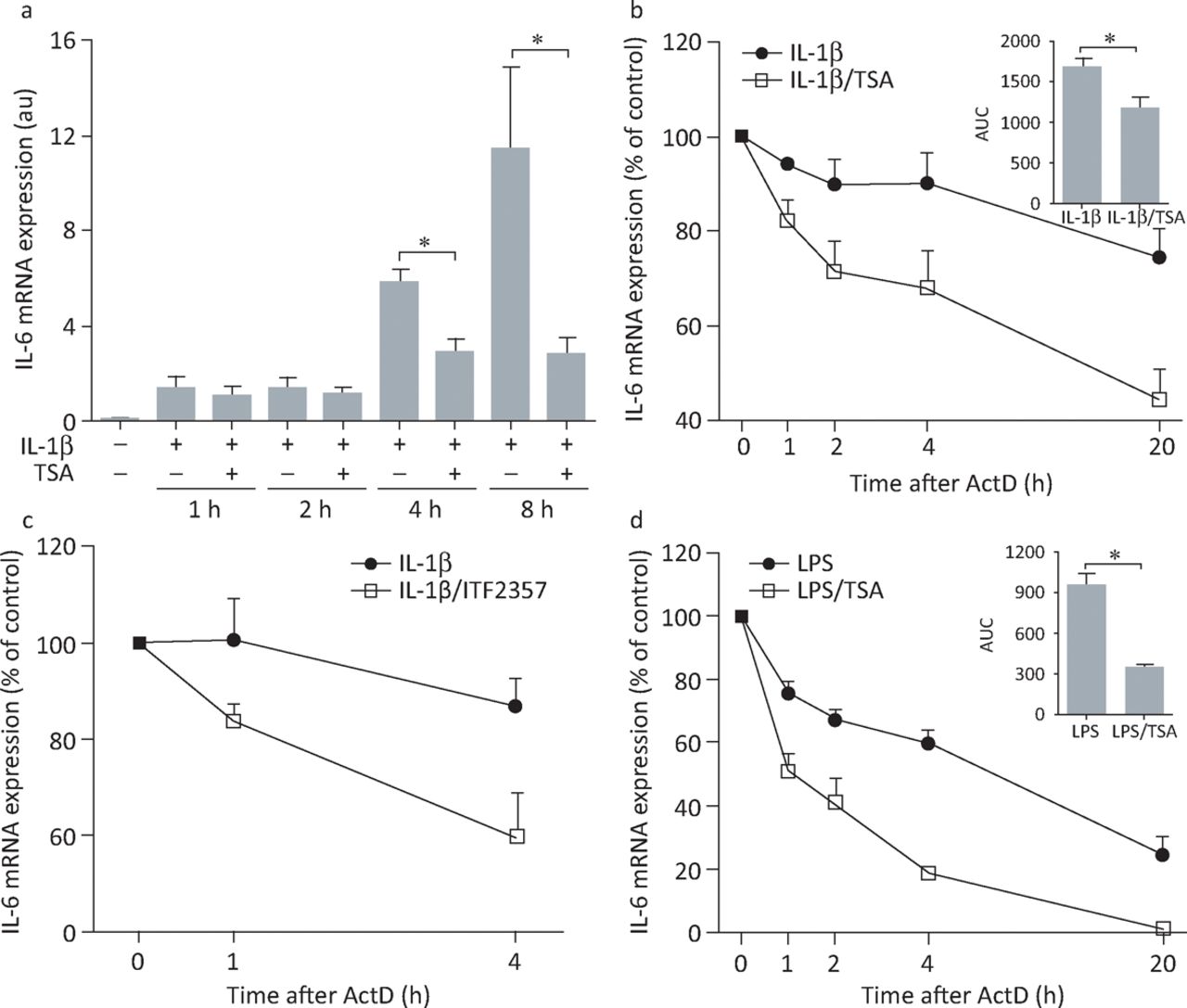
Histone deacetylase inhibitors accelerate IL-6 mRNA degradation in rheumatoid arthritis FLS and healthy donor macrophages. (A) FLS were left untreated or were treated with IL-1β (10 ng/mL) with or without 1 μM TSA, total RNA was extracted, complementary DNA was synthesised and temporal changes in IL-6 mRNA accumulation were monitored by quantitative polymerase chain reaction. Data represent the mean (± standard error) of five or six independent experiments. *p<0.05, Mann-Whitney U test. (B) FLS were stimulated with IL-1β (10 ng/mL) in the presence or absence of 1 μM TSA (n = 5) or (C) with IL-1β (1 ng/ml) in the presence or absence of 250 nM ITF2357 (n = 3). (D) Monocyte-derived healthy donor macrophages (n = 4) were stimulated with LPS (1 μg/ml) in the presence or absence of 1 μM TSA (n = 5). After 4 hours of stimulation, cells were washed and fresh medium containing 10 μg/mL ActD was added. RNA was extracted from the beginning of ActD treatment and the rates of IL-6 mRNA degradation in the presence or absence of histone deacetylase inhibitors were examined by quantitative polymerase chain reaction. Values for the 0 time point were normalised to 100%, and remaining values were expressed as the mean (± standard error) percentage of IL-6 mRNA concentrations compared with controls. For (B) and (D), the AUCs obtained for cells treated with an inflammatory stimulus alone or in the presence of TSA were calculated and differences between AUC values were analysed by the Mann-Whitney U test (insets, mean AUC ± standard error). *p<0.05. ActD = actinomycin D; AUCs = areas under the curves; FLS = fibroblast-like synoviocytes; IL = interleukin; LPS = lipopolysaccharide; TSA = trichostatin A. Reproduced with permission.17
Targeting readers: effects of BET bromodomain inhibition on FLS function and arthritis in vivo
Bromodomains are small protein interaction modules of approximately 110 amino acids that can bind to acetylated lysines on histone proteins. So far, 61 human bromodomains have been identified within 42 different proteins. Among these, the bromodomain and extra-terminal domain (BET) subfamily comprises four proteins, bromodomain-containing protein (BRD) 2, BRD3, BRD4 and BRDT, each of which possesses two tandem bromodomains. BRD2, BRD3 and BRD4 are expressed in most cells, but BRDT is found selectively in the testis.
Several potent small molecules that selectively bind to and inhibit the function of BET bromodomains have been developed. Recent studies have shown that these inhibitors have promise as potential therapeutic agents for the treatment of cancer, and increasingly, for autoimmune and inflammatory disease.
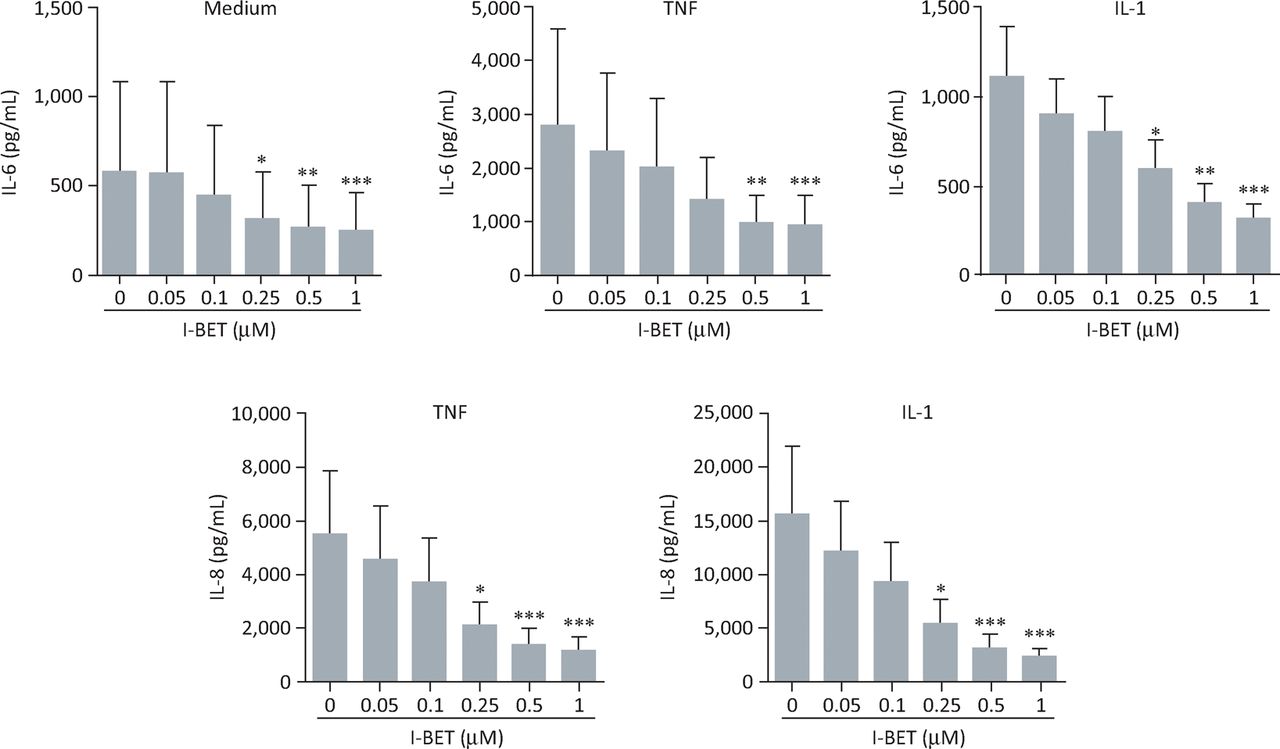
We assessed the effects of the BET inhibitor I-BET151 on the production of IL-6 and -8 by rheumatoid arthritis FLS.19 I-BET151 demonstrated a significant, dose-dependent inhibition of either spontaneous or cytokine-stimulated secretion of IL- 6 and -8 (Fig 3). I-BET151 inhibited cytokine-stimulated protein expression by the interleukins when added up to 4 hours after stimulation, suggesting that targeting of BET proteins can affect expression of these cytokines even after gene transcription has begun (Fig 4). Importantly, I-BET151 did not affect cell viability or interfere with cytokine signalling pathways.
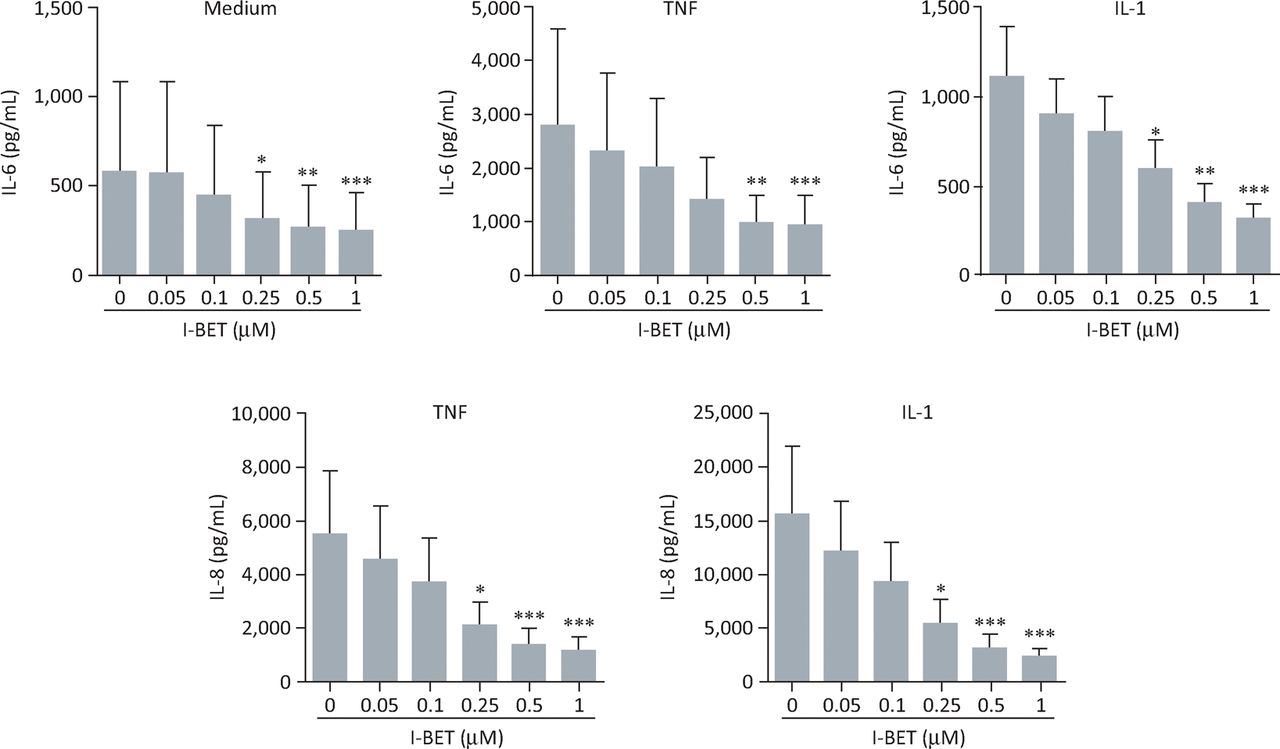
BET protein inhibition dose-dependently suppresses IL-6 and -8 production by rheumatoid arthritis FLS. Effects of i-BET151 on production of IL-6 (top panels) or -8 (bottom panels) by rheumatoid arthritis FLS cultured for 24 hours under basal conditions (medium alone), or in the presence of TNFα (10 ng/mL) or IL-1β (1 ng/mL). Data represent the mean (± standard error) for FLS obtained from 6 individuals. *p<0.05; **p<0.01; ***p<0.001 by analysis of variance with Bonferroni correction. BET = bromodomain and extra-terminal domain; FLS = fibroblast-like synoviocytes; IL = interleukin; TNFα = tumour necrosis factor α. Adapted with permission.19

I-BET151 can inhibit expression of IL-6 and -8 by rheumatoid arthritis FLS even if added after cells are exposed to inflammatory stimuli. I-BET151 was added to rheumatoid arthritis FLS cultures an hour before (prophylactic), an hour after (early) or 4 hours after stimulation of cells with TNFα (10ng/mL, left-hand panels), or IL-1β (1 ng/mL, right-hand panels). Data represent the mean (± standard error) for FLS obtained from six individuals. *p<0.05; **p<0.01; ***p<0.001 by analysis of variance with Bonferroni correction. Adapted with permission.19 BET = bromodomain and extra-terminal domain; FLS = fibroblast-like synoviocytes; IL = interleukin; TNFα = tumour necrosis factor α.
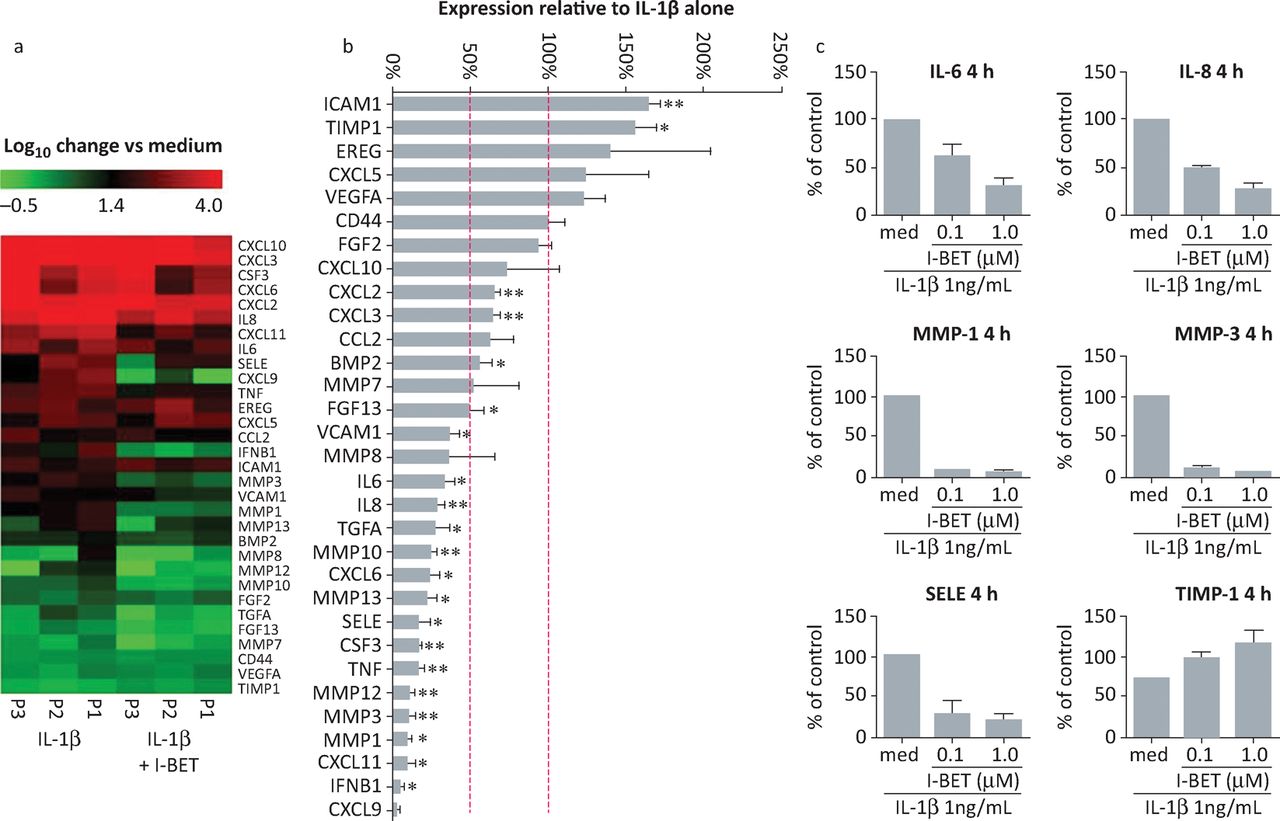
To investigate more broadly the effect of I-BET151 on rheumatoid arthritis FLS function, we assessed the effects on expression of a panel of genes linked to FLS pathogenetic behaviour (Fig 5). The results showed that I-BET151 substantially reduced mRNA concentrations for several genes linked to invasive and inflammatory properties in IL-1β-stimulated rheumatoid arthritis FLS. Several metalloproteinases, pro-inflammatory cytokines and chemokines were inhibited by I-BET151, which also enhanced the expression of some genes in the panel, consistent with published studies showing selective downregulation or upregulation of gene expression by BET inhibitors.20 Among the upregulated genes was tissue inhibitor of metalloproteinase (TIMP-1), showing that I-BET151-induced gene upregulation can also contribute to inhibition of FLS invasiveness.
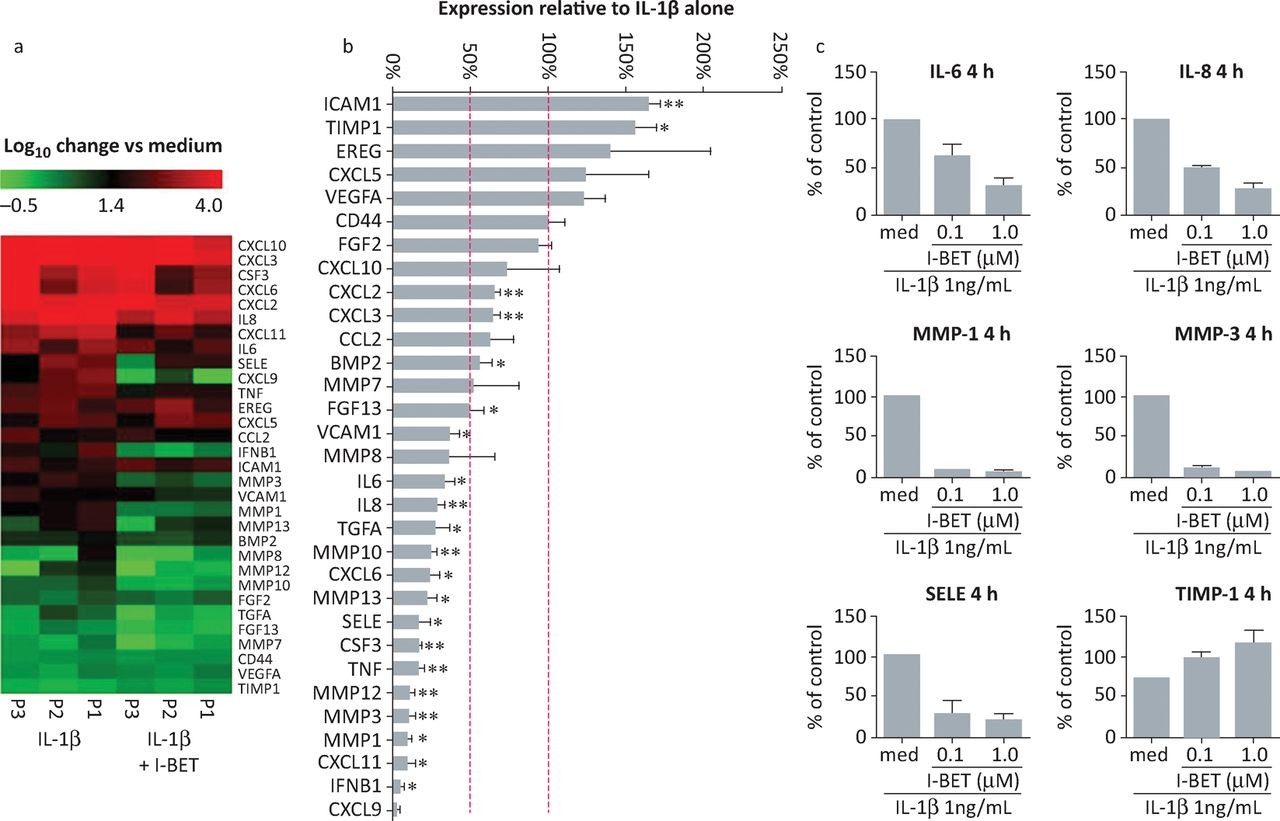
Selective modulation of rheumatoid arthritis FLS gene expression by i-BET151. (A, B) FLS from three different patients with rheumatoid arthritis were stimulated with IL-1β (1 ng/mL) for 4 hours in the presence or absence of 1 μM i-BET151. Total RNA was isolated, reverse transcribed and gene expression analysed by custom-designed quantitative polymerase chain reaction array. Original, log-transformed expression values relative to unstimulated cells are presented as heat maps in A. In B, data are presented as percentage of mRNA expression in I-BET151-treated cells compared with that in controls incubated with IL-1β alone. Dashed lines indicate expression levels equivalent to controls (100%) and 50% reduced compared with controls. *p<0.05; **p<0.01; ***p<0.001. (C) RASF (n = 2) were stimulated for 4 hours with IL-1β alone or in combination with 100nM or 1μM i-BET151. Total RNA was isolated, reverse transcribed and gene expression analysed by quantitative polymerase chain reaction array. Data are presented as percentage of mRNA expression in I-BET treated cells compared with that in controls treated with IL-1β alone. BET = bromodomain and extra-terminal domain; FLS = fibroblast-like synoviocytes; IL = interleukin; MMP = matrix metalloproteinase; RASF = rheumatoid arthritis synovial fibroblasts; SELE = selectin E; TIMP = tissue inhibitor of metalloproteinase. Adapted with permission.19
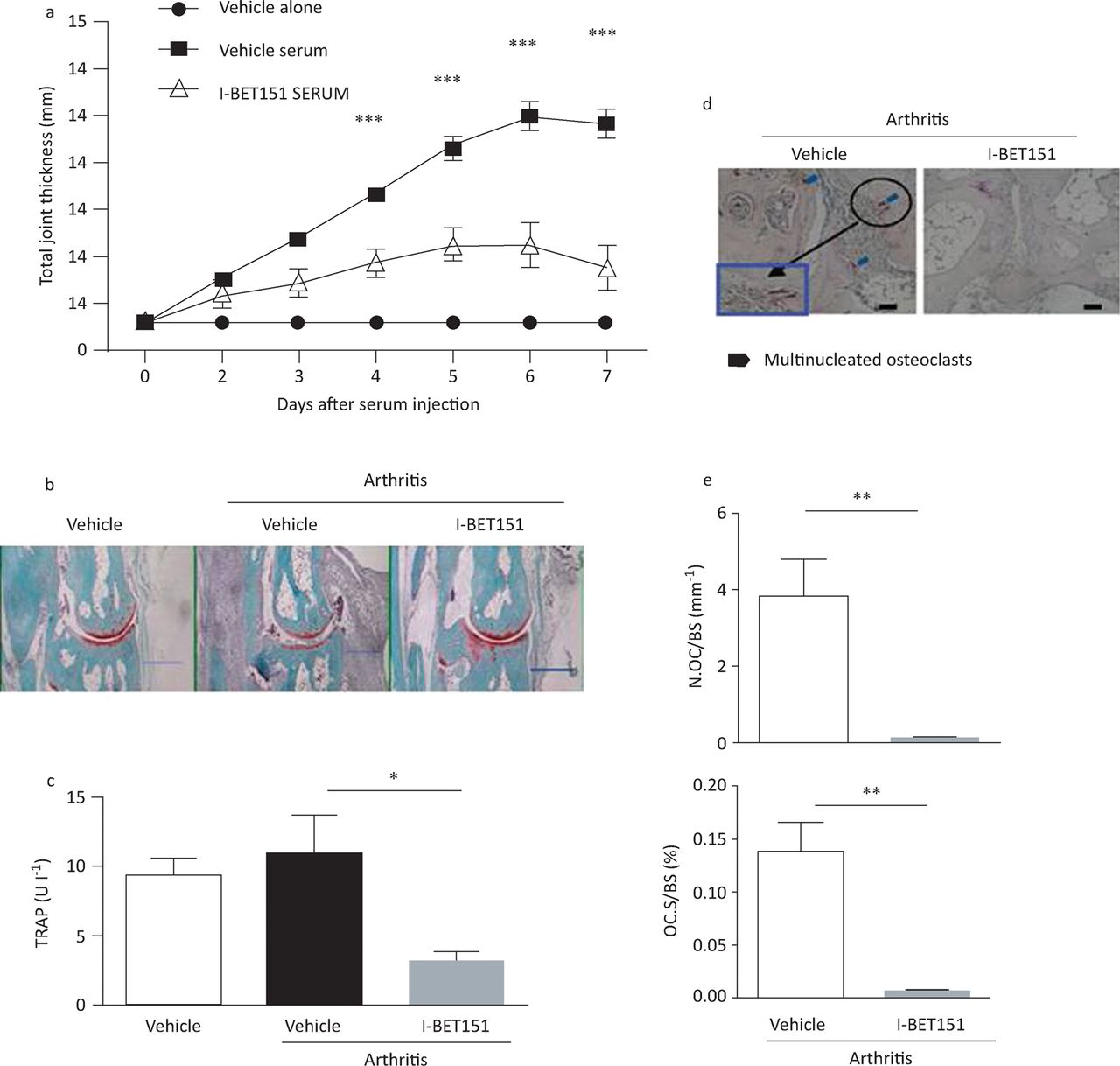
In view of these data showing that I-BET151 can inhibit the pro-inflammatory and invasive behaviour of isolated rheumatoid arthritis FLS in vitro, we investigated whether the compound could have broader effects on rheumatoid-arthritis-related mechanisms in a mouse model of arthritis21 – the K/BxN serum-induced arthritis model, in which transfer of serum from an autoimmune T-cell receptor transgenic mouse strain leads to arthritis in recipient mice characterised by joint inflammation and associated bone resorption.22 Joint swelling and inflammation and bone resorption were substantially less in mice treated with I-BET151 than in untreated controls (Fig 6). Furthermore, treatment led to reduced numbers of osteoclasts in periarticular bone (Fig 6c–e), which is linked to I-BET151’s ability to directly inhibit osteoclast differentiation.21 Taken together, these data suggest that BET inhibitors can target several mechanisms associated with the pathogenesis of rheumatoid arthritis.

I-BET151 attenuates K/BxN serum-induced arthritis. Arthritis was induced in C57BL/6 mice by intraperitoneal injection of 100 μL of K/BxN serum on days 0 and 2. Arthritic mice were divided into two groups (vehicle versus I-BET151; n = 9 mice per group) and were administered vehicle or i-BET151 (40 mg/kg) once on days 0 and 2 and twice daily from day 3 to day 6. The control group (n = 9) only received vehicle but not arthritic serum. (A) Time course of joint swelling in the presence and absence of i-BET151 treatment. Values are the mean (± standard error) of six mice per group. ***p<0.001 by two-way analysis of variance (B) Histology of ankle joints of mice examined 11 days after the first arthritogenic serum injection. Hind paws were fixed and decalcified, and ankle joint sections were prepared and stained with haematoxylin, fast green and Safranin O. Scale bar: 500 mm. (C) TRAP concentrations were measured 7 days after arthritis induction. *p<0.05 by two-way analysis of variance. (D) TRAP staining of histological sections obtained from control or i-BET151 treated arthritic mice. Blue square (inset) shows an enlarged image. Scale bar: 100 mm. (E) Histomorphometric analysis. All data are shown as mean ± SEM **p<0.01 by t-test. BET = bromodomain and extra-terminal domain; N Oc BS–1 = osteoclast number per bone surface; OcS BS–1 = osteoclast surface area per bone surface; SEM = standard error of the mean; TRAP = tartrate-resistant acid phosphatise. Reproduced with permission.21
Concluding remarks
There is strong evidence for the key role of FLS in driving the progression of rheumatoid arthritis, but available treatments do not target these cells. The finding that these cells show an imprint of patients’ heterogeneity is an indication that FLS undergo epigenetic changes in response to the inflammatory environment of the synovium in rheumatoid arthritis. This notion is supported by a small study, which showed that the methylome of early rheumatoid arthritis had a pattern distinct from that of long-standing disease.23 Such epigenetic changes in established disease can explain why the so-called therapeutic window of opportunity for current treatments is limited to early rheumatoid arthritis.24 Interfering with epigenetic pathways – for instance, by inhibition of histone deacetylase or BET bromodomains – could therefore represent a novel therapeutic approach in the treatment of chronic immune-mediated inflammatory disease in rheumatoid arthritis and beyond.
- © Royal College of Physicians 2015. All rights reserved.
References








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.