
ABSTRACT
Idiopathic pulmonary fibrosis (IPF) is characterised by progressive accumulation of scar tissue in the lung and is associated with a median life expectancy of 2–4 years. Until recently, treatment options were limited, focusing on ineffective anti-inflammatory therapy, palliation, transplant or trial recruitment. Significant recent advances in the field have led to two novel anti-fibrotic agents, pirfenidone and nintedanib, which have been shown to significantly slow disease progression in IPF. This article outlines the approach to management of IPF, the role of specialist centres and specialist interstitial lung disease multidisciplinary review, and explores both the trial evidence and practical considerations in the use of these anti-fibrotic agents.
Background
Idiopathic pulmonary fibrosis (IPF) is a chronic fibroproliferative disease exclusively affecting the lungs. It is characterised by the irreversible accumulation of scar tissue that gradually replaces normal lung parenchyma, and leads to symptoms of exercise intolerance and dyspnoea, with a median life expectancy of 2–5 years. IPF has been classified as a rare disease but evidence from national UK databases indicates that the incidence is increasing. At present there are estimated to be between 4.6 and 8.8/100,000 new cases per year in the UK and a prevalence of 28/100,000; however this may be an underestimation as a recent US study examining Medicare claims over 10 years found a far higher prevalence of 93.7/100,000.1–3
By definition, the aetiology of IPF is unknown but the pathogenesis is thought to be part genetic predisposition and part environmental. Environmental risk factors include smoking and exposure to wood and metal dusts. Possible disease triggers such as viruses and gastro–oesophageal reflux (GOR) have been investigated and appear to be associated with IPF in a subset of patients, although direct causality has not been proven. More recently the focus has switched to examining the genetic contribution, following the identification of specific mutations found in familial forms of pulmonary fibrosis. Mutations in genes encoding telomerase and surfactant, and polymorphisms in the MUC5B gene among others, have shed light on pathogenic pathways and led to the discovery that the frequencies of these mutations are significantly higher in the IPF population. Indeed, a familial link in patients with IPF is seen in between 2 and 20% of cases.3 Age is an important risk factor and it has been hypothesised that this may relate to the defects in cellular repair due to age-related shortening of telomeres.4
While the exact pathogenic mechanisms of IPF remain elusive, there have been significant advancements in understanding the nature of the disease and some of the dysregulated processes that occur. IPF is not a chronic inflammatory disease (as once hypothesised) but a disease characterised by a fibroproliferative response resulting from an aberrant response to injury; minor, but probably repetitive, injury to the alveolar epithelial cells triggering a cytokine cascade leading to the recruitment, proliferation and activation of cells involved in the healing response. Activated fibroblasts known as myofibroblasts are the main effector cells in IPF responsible for the laying down of extracellular matrix. Key mechanisms to switch off the healing response are in some way deficient and the accumulation of abnormal extracellular matrix may drive a profibrotic positive feedback loop resulting in the ongoing deposition of fibrotic tissue.5–8
Until recently, the only real treatment options available were that of palliation, trial recruitment or referral for lung transplantation. The majority of earlier clinical trials utilised drugs with isolated molecular targets, such as tumour necrosis factor-α, interferon-γ and endothelin receptor inhibition, and met with failure; most probably because of the multitude of cellular processes that are involved in IPF pathogenesis. The field has been significantly advanced with the development of novel therapies, such as pirfenidone and nintedanib, which are pleiotropic in their anti-fibrotic activities, and have been shown to significantly slow disease progression. While a cure is still elusive, these drugs provide benefit and hope to many patients with this devastating disease.
The aim of this review is to provide an overview of current IPF management strategies with a focus on both symptom-directed and disease-directed therapeutic agents.
Defining the diagnosis of IPF
Until 15 years ago, little focus was placed on IPF and in the UK many specialists referred to the disease as cryptogenic fibrosing alveolitis. This corresponded to a clinical pattern similar to that seen in patients with IPF but included other fibrotic lung diseases within the cryptogenic fibrosing alveolitis ‘umbrella’, such as chronic hypersensitivity pneumonitis and fibrotic non-specific interstitial pneumonia (NSIP).9,10 The realisation that defining the type of interstitial lung disease (ILD) more carefully had both important management and prognostic implications (with NSIP typically pursuing a more indolent course), led to the understanding that IPF had a particularly poor prognosis, similar in fact to lung cancer. In 2002, the American Thoracic Society (ATS)/European Respiratory Society (ERS) committee thus redefined the diagnostic criteria for IPF,11 and guidelines for diagnosis and management were refined in 2011 by the ATS/ERS/Japanese Respiratory Society (JRS)/Latin American Thoracic Association (ALAT).12 In addition to classical clinical features, the appearance of a usual interstitial pneumonia (UIP) pattern on either histological examination (Table 1) or high-resolution computed tomography (HRCT) imaging (Table 2) was necessary to provide a secure diagnosis. As a UIP pattern is not unique to IPF and can be associated with known causes or conditions, a careful history and examination during initial evaluation is essential. Connective disease-associated ILD (most commonly rheumatoid arthritis), drug toxicity, chronic hypersensitivity pneumonitis, sarcoidosis and asbestosis can all cause a UIP pattern of fibrosis, and management of each disease differs significantly. Screening blood tests include antinuclear antibody, rheumatoid factor and anti-cyclic citrullinated peptide, with possible others as dictated by the clinical picture.9,12
The histopathological criteria for a diagnosis of UIP (adapted from the ATS/ERS/JRS/ALAT statement for IPF 2011. Reprinted with permission of the American Thoracic Society.12 Copyright © 2016 American Thoracic Society. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
The radiological criteria for a diagnosis of UIP (adapted from the ATS/ERS/JRS/ALAT Statement for IPF 2011. Reprinted with permission of the American Thoracic Society.12 Copyright © 2016 American Thoracic Society. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
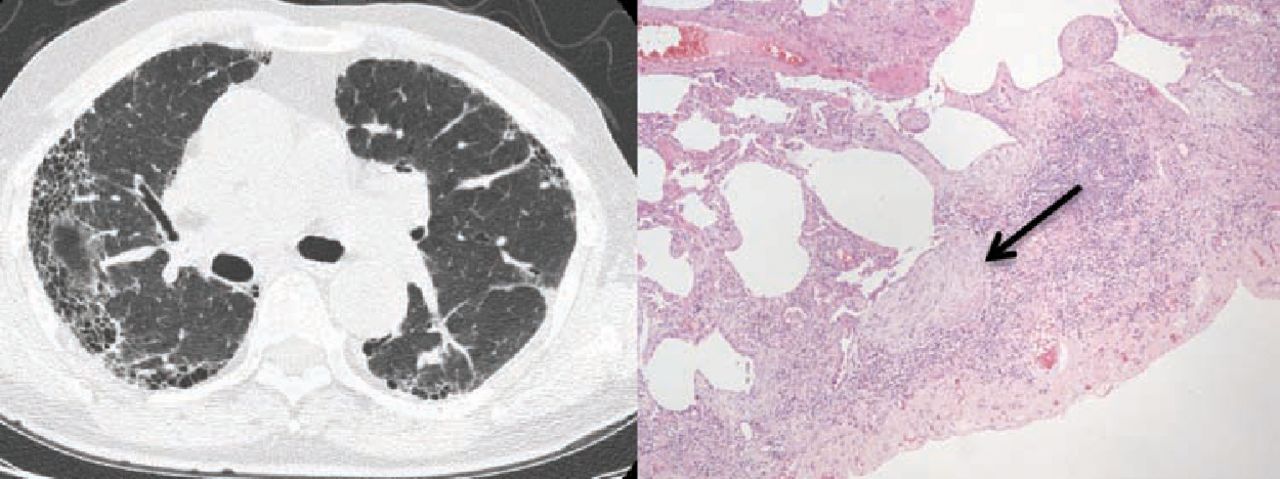
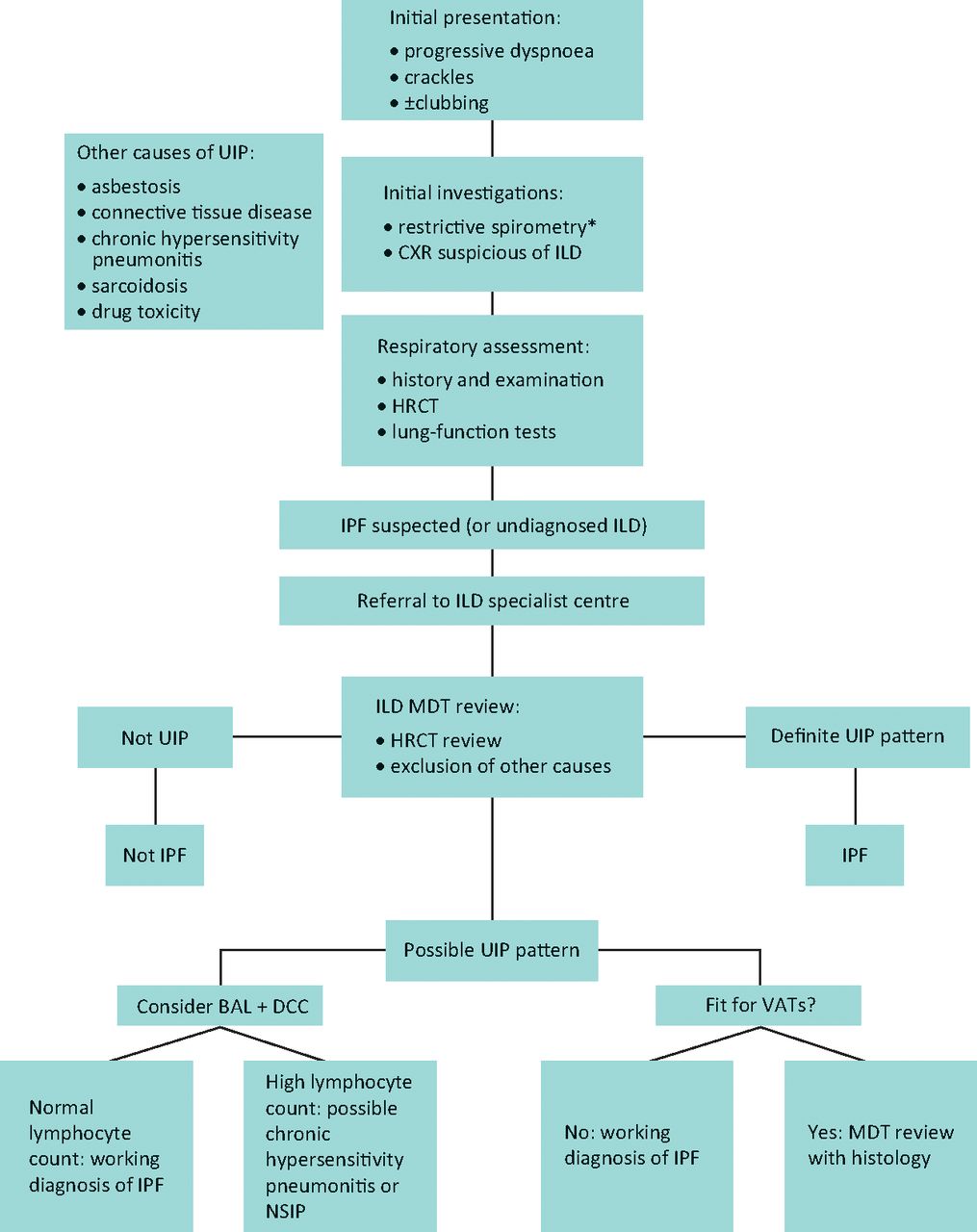
The presence of a definite usual interstitial pneumonia pattern on HRCT, with subpleural basal honeycombing, traction bronchial dilatation and reticulation, in a patient without other known causes of UIP, enables the specialist clinician to make a robust diagnosis of IPF (Fig 1a). In a significant proportion of patients however, these typical CT appearances are not present, most usually an atypical distribution of disease or absence of clear honeycombing. Recent guidelines, including the National Institute for Health and Care Excellence (NICE) clinical guidelines published in 2013,13 acknowledge the diagnostic difficulty that often occurs and recommends that all suspected cases of IPF be reviewed by a multidisciplinary team (MDT) with expertise in ILD, including a consultant radiologist, histopathologist and specialist ILD chest physician (Fig 2). In the UK, specialist centres for ILD with concentrated expertise have been provisionally identified as part of the specialist commissioning process, with many centres operating in hub-and-spoke arrangements with regional referring centres. A thoracic surgeon should also form part of the MDT, as a surgical lung biopsy (video-assisted thoracoscopic surgery) is an option for patients that are able to tolerate the procedure; there is however a defined morbidity and mortality for the procedure in an increasingly elderly population.13 Lung biopsies in IPF show the features of temporal and spatial heterogeneity, and the pathognomonic feature of UIP, the fibroblastic focus (Fig 1b). Chronic hypersensitivity pneumonitis is a known cause of a UIP pattern on CT, and can be further investigated with bronchoalveolar lavage for differential cell count; a lymphocytosis is supportive of chronic hypersensitivity pneumonitis.

(a) CT appearance of a definite UIP pattern, with subpleural basal reticulation, traction bronchial dilatation and honeycombing; (b) lung biopsy appearance in UIP – temporal and spatial heterogeneity and fibroblastic foci. UIP = usual interstitial pneumonia.
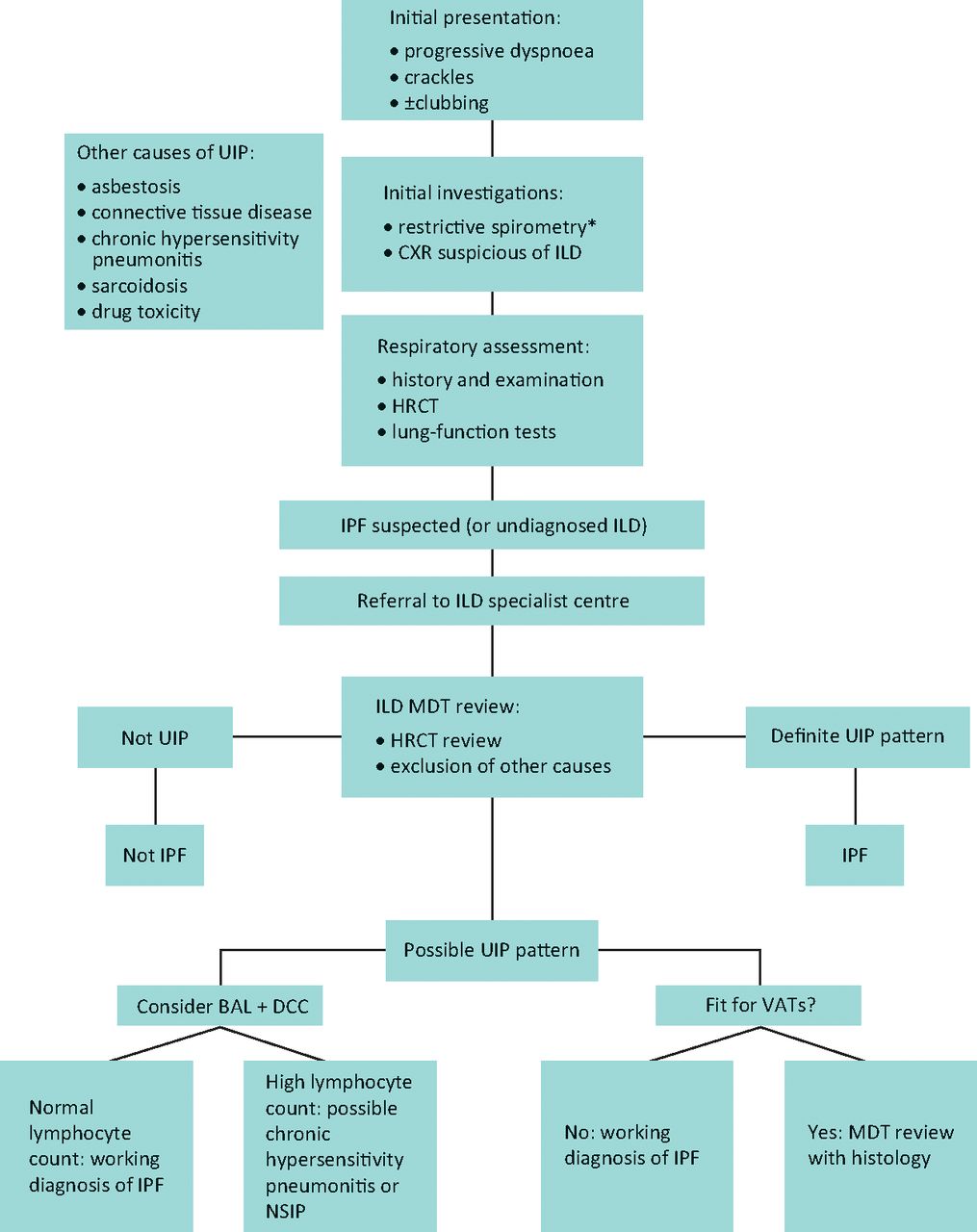
The idiopathic pulmonary fibrosis diagnostic pathway. *Spirometry may not be restrictive in early disease or when coexistent airways disease is present. BAL = bronchoalveolar lavage; CXR = chest X-ray; DCC = differential cell count; HRCT = high-resolution computed tomography; ILD = interstitial lung disease; IPF = idiopathic pulmonary fibrosis; MDT = multidisciplinary team; NSIP = non-specific interstitial pneumonitis; UIP = usual interstitial pneumonitis; VATS = video-assisted thoracoscopic surgery.
A diagnosis of IPF made via MDT consensus, in other words a ‘working diagnosis of IPF’, has become even more relevant in this new era of anti-fibrotic drugs licensed solely for IPF. Patients without classical features of ‘definite’ UIP on CT, and who are unsuitable for biopsy, risk being deprived of optimal management without specialist input. Interestingly, a study by Fell et al examined the clinical and radiological characteristics of patients who had biopsy-proven UIP and found a high likelihood of having IPF in those who were aged over 75 with moderate fibrosis on HRCT. While this study requires further validation, it indicates that advancing age and fibrotic CT changes are strongly predictive of IPF in patients without predisposing causes.14
Overall management principles
Despite the recent focus and advances in the pharmacological management of IPF, drug treatment is only a component of a patient's care in clinical practice. As with many chronic diseases, the healthcare needs of a patient must be addressed through an integrated approach to care involving the ILD specialist team. As a minimum, this consists of a consultant physician with ILD expertise, respiratory physiotherapist, nurse specialist and pharmacist. Over time, the health-related requirements of the patient typically increase and supportive and symptomatic management becomes the priority. Close liaison with the general practitioner (GP) through all stages of the disease is essential, and involvement of community nursing and palliative care teams are helpful, especially during the later stages of illness.
Approaches to management
When a patient is first diagnosed (via ILD MDT consensus), there are a number of management strategies that can be adopted depending on the stage of the disease, how quickly it is progressing, the health status, and preferences of the patient. More than one approach is often necessary and the management options highlighted below are not mutually exclusive. All appropriate treatment options should be discussed early following the diagnosis of IPF to enable patients to make informed decisions.
Watch-and-wait approach
Patients presenting with early disease, or who have combined emphysema and IPF, may have preserved lung function tests and therefore not qualify for anti-fibrotic therapy under NICE guidance, which currently advocates therapy if the forced vital capacity (FVC) is between 50 and 80% of predicted value. Other patients may have minimal symptoms and wish to delay treatment due to concerns about the impact that potential side effects could place on their quality of life. In these patients a careful monitoring strategy is advised and regular lung-function tests every 3–6 months enable the clinician to detect evidence of worsening disease. A fall in FVC of >10% or transfer factor for carbon monoxide (TLCO) of >15% is representative of significant disease progression and analysis of clinical trial data revealed that a decline in FVC of >10% over 6 months was associated with 5-fold risk of mortality over the subsequent year.15 Furthermore, recent data suggest that even smaller decreases in FVC of 5% are clinically significant and stratify patients into a poorer prognostic group,16 further emphasising the need for careful monitoring.
Active disease-directed treatment
In the UK at the time of writing, active treatment is currently restricted to pirfenidone, although it is anticipated that nintedanib will gain NICE approval for use in 2016, providing a therapeutic alternative. NICE guidance has restricted the use of pirfenidone in the UK to patients with an FVC of between 50 and 80%. Patient selection is key; we advocate anti-fibrotic therapies for patients in whom IPF is the predominant respiratory pathology, as the potential benefits are significantly attenuated in the context of significant emphysema, pulmonary hypertension or cardiac comorbidity. Frailty and reduced BMI appear to be associated with increased risks of side effects. Patients are monitored carefully while on therapy by both the specialist ILD team (often in a shared care arrangement with the referring secondary care centre) and GP, and lung function tests are measured during the course of treatment. A fall in FVC of >10% after a year of pirfenidone therapy is considered to represent treatment failure under NICE guidance and is an indication to stop the drug in the UK. It is very likely that similar prescribing constraints will apply to nintedanib.
Symptom-based approach
A subset of patients present with advanced disease or have extensive comorbidities that preclude the use of anti-fibrotic therapy. In these patients, regular review and lung function monitoring remains important as it enables the clinical team to ensure that their medical management is optimised and reversible causes of deterioration are addressed. Symptomatic relief of breathlessness and supportive care are essential aspects of management in all patients throughout their illness. Oramorph and lorazepam are often effective for dyspnoea control, as are pacing and breathlessness control exercises, and low-dose corticosteroids or codeine may be useful for intractable cough. In those with rapidly progressive or advanced disease, a frank discussion with the patient and their carers regarding end-of-life planning may be appropriate, in conjunction with palliative care involvement.
Pharmacological treatment options
The treatment recommendations for IPF have changed substantially over the past seven years. The 2008 British Thoracic Society guidelines recommended triple therapy with prednisolone, azathioprine and N-acetylcysteine (NAC); however following the PANTHER-IPF trial, this treatment approach was quickly withdrawn in view of reported increased hospitalisation and mortality in the active arm.9,17 The 2011 ATS/ERS/JRS/ALAT guidelines adopted the somewhat challenging GRADE methodology in their recommendations, but due to the overall lack of robust evidence at that time for any effective therapies, the take-home message from the guidelines was that of best supportive care in conjunction with clinical trial recruitment where possible.12 The publication of the pirfenidone data soon afterwards provided renewed optimism and the review of the drug by the NICE Technology Appraisal in 2013 resulted in recommendations for its use in patients with mild-moderate disease.18 Nintedanib has since been approved by the European Medicines Agency and is licensed in Europe for all patients with IPF regardless of severity. It is currently under review by NICE and prescribing approval is anticipated in the UK in 2016.19 The 2015 update of the ATS/ERS/JRS/ALAT guideline, published July 2015, conditionally recommends the use of pirfenidone and nintedanib for IPF, and consideration for anti-acid therapy.20
Pirfenidone
Pirfenidone is a novel, orally available anti-fibrotic drug approved for use in the UK for patients with mild-moderate IPF. Its exact mechanism of action is unknown, but in a murine model of fibrosis administration of pirfenidone led to a reduction in the production of key profibrotic cytokines including transforming growth factor-β, interleukin-1β and fibroblast growth factor (FGF). A decrease in lung collagen content and fibrosis scores was demonstrated and proliferation of fibroblasts was attenuated, indicating that pirfenidone acts by inhibiting important fibrogenic pathways.
The development of pirfenidone for clinical use follows a decade of clinical trial work involving the recruitment of over 1,700 patients into five trials designed to test its safety and efficacy. Initial phase-II and -III trials were undertaken in Japan and showed that pirfenidone significantly reduced the decline in FVC over a year compared to placebo.21 To evaluate this further, two replicate randomised controlled trials were undertaken; the CAPACITY trials recruited patients in the US, Europe and Australia to receive high or low-dose pirfenidone or placebo. The trials spanned a 72-week period and the primary outcome – change in FVC from baseline – was reached in one but not both of the studies.22 When the data from both trials were pooled (in pre-specified analysis), the primary endpoint was achieved in patients receiving high-dose treatment, alongside the secondary endpoints of reduction in decline of six-minute walk test (6MWT) distance and prolonged progression-free survival time. On the basis of these findings the European Commission approved pirfenidone in 2011. In the USA, driven by the Food and Drug Administration (FDA), the ASCEND trial recruited 555 patients to receive active treatment or placebo, and the results of the study were published in 2014.23 Disease progression was reduced in the pirfenidone arm as evidenced by reduction in the mean decline in FVC by 195 mL compared to placebo. Furthermore, pooled data analysis from the CAPACITY and ASCEND trials demonstrated a reduction in both all-cause and IPF-related mortality. The FDA subsequently fast-tracked the approval of pirfenidone for clinical use awarding it ‘breakthrough therapy’ status.
Adverse events reported in the pirfenidone arm were common in the trials and gastrointestinal side effects were the most frequently reported. Nausea, anorexia, dyspepsia and diarrhoea were more common in the treatment arm, as were elevations in aspartate transaminase and alanine transaminase. Photosensitivity reactions were also more common and experienced by 12%, compared with 1.7% of participants treated with placebo. Overall however, side effects were considered mild or moderate, and only a small number had to discontinue the drug. The RECAP trial is an open-label, extension trial looking at the long-term safety of pirfenidone. Interim data show that the side-effect profile is similar to that seen in the clinical trials and confirms that pirfenidone is generally well tolerated.24
Nintedanib
Nintedanib is an intracellular triple tyrosine kinase inhibitor that binds competitively to receptors to vascular endothelial growth factor, platelet-derived growth factor and FGF, blocking downstream signalling pathways. These growth factors are recognised mediators of fibrogenic pathways and, as with pirfenidone, the administration of nintedanib following bleomycin lung injury in mice results in reduced inflammation and fibrosis.25 The inhibition of fibroblast function is thought to be the main mechanism by which nintedanib modulates the disease processes in IPF.
The safety and efficacy of this agent have been evaluated in three clinical trials. The first study, (TOMORROW) was a phase-IIb dose-finding study that recruited 432 patients with mild-moderate disease.26 Patients received either placebo or four different doses of nintedanib for 52 weeks; in the highest dose group a reduction in the decline of FVC was noted. Thereafter followed two replicate trials recruiting 1,069 patients with IPF from 24 countries; patients who were enrolled into IMPULSIS-1 and IMPULSIS-2 trials received either maximum-dose nintedanib (150 mg bd) or placebo for one year; similar to the pirfenidone trials, the rate of decline in FVC over a year was the primary outcome.26 Both trials met the primary outcome of slowing FVC decline and in IMPULSIS-2, but not in IMPULSIS-1, the secondary outcomes of time to first IPF acute exacerbation (IPF-AE) and change in health-related quality-of-life scores were also met.26 While the pooled data from both trials did not show a significant difference in the IPF-AE endpoint, a post-trial adjudicated review of the investigator-reported events revealed that after removing known causes of deterioration such as pulmonary embolism and left ventricular failure, the numbers of acute exacerbations in the placebo group were significantly higher. The FDA also awarded nintedanib ‘breakthrough therapy’ status in 2014.
Importantly, the criteria for the enrolment of patients into the IMPULSIS studies were less stringent than that used previously. A definite diagnosis of IPF was not required but rather the changes needed to be ‘consistent’ with IPF. Patients with coexistent emphysema were also included in the trial, resulting in a far more heterogeneous study population than that seen previously. While this approach could be criticised for the possible diagnostic inaccuracies that incurred, leading to the inclusion of patients who had a fibrotic lung disease other than IPF, the study design can be also commended for more closely replicating the reality of management in clinical practice. Importantly, no outcome differences were seen between patients with ‘definite IPF’ and a ‘working diagnosis of IPF/probable IPF’.27
A similar proportion of serious adverse events occurred in treatment and placebo arms (around 30%) and the most common adverse effect in the nintedanib arm was diarrhoea, which was reported by 62 versus 18% of patients. Cases however were mostly either mild or moderate, and only 4% of patients discontinued the drug because of this side effect. Diarrhoea was reported if one episode was reported over one year. Nintedanib is not recommended in patients with moderate or severe liver impairment and should be used cautiously in those with increased bleeding risk, including those on anticoagulants.
Managing side effects of pirfenidone and nintedanib
Side effects from anti-fibrotic therapy tend to be most pronounced during the initiation and titration period, and therefore close supervision and support is particularly important during this process to ensure successful compliance. The ILD specialist nurse plays an essential role in the management of patients commenced on active treatment, and careful counselling prior to starting medication helps prepare patients for potential side effects. The ILD nurse also acts as a point of contact for patients, providing telephone support for medication and symptom queries, and liaises with the medical team when appropriate. Regular clinic appointments including provision for ‘emergency clinic slots’ are necessary to ensure that problematic side effects are managed promptly and appropriately.
Pirfenidone
Nausea and vomiting are fairly common initial side effects which can be helped by taking the medication with food and eating smaller but more frequent meals. Antiemetics can also be helpful as a temporary adjunct. Diarrhoea can be treated symptomatically with loperamide provided other causes have been excluded. If these measures are unsuccessful, temporary dose reduction followed by slow reintroduction of pirfenidone may be necessary. Loss of appetite is a relatively common problem requiring dietary advice and focus on high-calorie foods. A temporary reduction may also be effective in improving symptoms, but significant and ongoing weight loss may be an indication to halt therapy. Tiredness and fatigue are potential side effects and lifestyle and sleep hygiene advice may be helpful. If this fails, short-term dose reduction may be necessary. In view of the skin photosensitivity caused by pirfenidone, patients need to be advised to wear factor 50 SPF sunscreen, and to cover up when exposed to high-level UV light. Rash is a possible, though less common, complication if this advice is followed. Skin erythema may respond to emollients; however a rash requires interruption of therapy.
Nintedanib
The main potential side effect of nintedanib is diarrhoea, which in most cases is mild to moderate and improves over time; although there are very rare reported instances of diarrhoea with involuntary incontinence. In many centres, loperamide is co-prescribed initially alongside the drug as required. If diarrhoea persists, a temporary drug holiday may be required. Nausea, vomiting and abdominal discomfort are possible side effects, and are managed symptomatically, with dietary advice or co-prescription of antiemetics. Liver function abnormalities are less common, and rigorous attention to other hepatotoxic medication is required. If these measures are not effective, dose reduction to 100 mg bd is an option.
Anti-fibrotic selection
At the time of writing pirfenidone is the only NICE-approved active treatment option for IPF in the UK; however nintedanib is anticipated to be approved by NICE for use within the NHS in 2016. The efficacy profile of pirfenidone and nintedanib are similar, with equivalent slowing of disease progression; the decision regarding which treatment to commence requires careful consideration. Currently, there are more data on the long-term effects of pirfenidone, and while significant mortality benefits are becoming apparent, it is currently unknown whether similar findings will be seen with nintedanib as data emerge from extension studies. Although only apparent in the pooled study comparison, a trend toward an impact on IPF-AE is limited to nintedanib. Financial considerations are certain to play a role in prescribing practices and the comparative costs of the drugs are not yet clear, although are likely to be reasonably similar. Restrictions on FVC (50–80%) dictate which patients are currently eligible for pirfenidone; it is unknown whether the same restrictions will apply to nintedanib, although provisional comments from NICE suggest that the same criteria may apply (this guidance may change with the final NICE technology appraisal recommendations for nintedanib). There is an urgent clinical need for anti-fibrotic use with mild disease in order to prevent decline, however drugs are considered less cost effective by NICE (on health economic grounds) with mild disease.
If the prescribing criteria are the same for both drugs then practical considerations are likely to determine the therapeutic strategy. Side effects, if present, may be manageable with one drug but not the other; however it is unlikely to be predictable which response a patient will have to either of the treatments, and a trial and error approach may need to be adopted. Lifestyle considerations should not be underestimated and the tablet burden of pirfenidone versus nintedanib (9 vs 2 tablets/day) is a consideration, although the dosing regime with pirfenidone also allows a more gradual drug titration to side effects if necessary. The fastidious daily application of sunscreen to prevent phototoxicity is likely to be a significant disincentive in a proportion of patients considering treatment with pirfenidone. Diarrhoea is the major drawback for nintedanib, and patients need to be made aware of the very small risk of involuntary diarrhoea. In addition, as nintedanib potentially increases the risk of bleeding, it should be avoided in patients with bleeding tendencies, those taking anti-coagulants and patients planning major surgery. There is a lack of data for the use of nintedanib for patients on an active transplant list, with international and regional variation in practice, and local discussions with the transplant referral centre in advance of prescription are advised. Pirfenidone has been in use for a longer time period, hence familiarity, however most specialist centres have used nintedanib as part of an individual patient access scheme and are now equally cognisant of the practical considerations of both agents.
Other considerations
Comorbidities
In addition to the strategies highlighted above, the management of comorbidities is an essential component of clinical care. IPF is a disease affecting mainly elderly patients and breathlessness is often multifactorial. Ischaemic heart disease and heart failure are common causes for breathlessness and should be investigated when suspected clinically. An echocardiogram is useful to look for evidence of left ventricular dysfunction and pulmonary hypertension. A disproportionate increase in right ventricular pressure in a patient with early disease may warrant further investigation and referral to a specialist in pulmonary hypertension. Patients may also have coexisting airways disease and inhaler therapy should be optimised. Smoking cessation should be offered in those continuing to smoke. Causes of chronic cough besides ILD need to be considered, such as post-nasal drip and gastro–oesophageal reflux, and trials of appropriate treatment initiated.
Early referral for transplant
The 2011 ATS guidelines strongly recommend early referral for lung transplantation in appropriate patients. In the US, IPF is one of the most common indications for transplant and five-year survival after transplant is estimated to be between 50 and 56%.10,28 The NICE guidelines also advocate that the option of lung transplantation is discussed with patients without absolute contraindications within 3–6 months of diagnosis. Absolute contraindications to transplant as determined by the NHS Blood and Transplant Authority include: recent cancer (within the past five years), those in an unstable critical condition, poor compliance with medication, substance dependency (including drug, alcohol and tobacco), those with untreatable psychiatric conditions or a poor social support network, and patients with advanced dysfunction in other organs. Anatomical chest and spinal deformity may also impede access at surgery and prevent optimal ventilation postoperatively, and can be an absolute contraindication. Relative contraindications form a long list but include age >65 years, a BMI >30 or <17, osteoporosis, steroid dependency (>15 mg/day of prednisolone), HIV and hepatitis B or C infection, and those on mechanical ventilation.28
Pulmonary rehabilitation
Pulmonary rehabilitation (PR) has been long established as an effective treatment strategy in chronic obstructive pulmonary disease (COPD) and has been shown to improve 6MWT distance and wellbeing. Based on these findings, PR has been recommended by NICE in the management of IPF. A recent Cochrane review analysed nine studies investigating the effect of PR on patients with different types of ILD, and concluded that PR was both safe and resulted in improvements in functional exercise capacity, dyspnoea and quality of life immediately following the course. These findings were extended to participants with IPF and a significant improvement was noted in 6MWT distance following exercise. The numbers of participants in these studies however were small and, due to the different reporting methods, the quality of evidence was rated low to moderate.30 The long-term effects of PR within this population group are uncertain and a recent study by Jackson et al found that while IPF patients allocated PR showed improvements in quality of life and physical activity, these changes were only noted for the duration of the intervention and not three months later.31 The lack of sustained improvements may in part reflect the progressive nature of the disease but it is also possible that the rehabilitation needs of some IPF patients differ from those with other chronic lung diseases, with disease severity playing an important part in treatment response. As recommended by the NICE guidelines, further trials are required involving IPF patients with differing disease severities to look at the effect of both short- and long-term outcomes.32,33
Oxygen therapy
The IPF NICE guidelines advocate the regular monitoring of oxygen saturations in patients with IPF both at rest and during exertion, with the recommendation that oxygen should be offered to all those that show evidence of significant desaturation. These recommendations are in keeping with the oxygen prescribing guidelines for patients with COPD, which is not surprising given that the evidence for the use of oxygen for IPF is derived entirely from studies analysing the effect of oxygen in patients with COPD. While a Cochrane review into the use of oxygen in COPD reported a mortality benefit for the use of long-term oxygen therapy in those with hypoxia, it is unclear whether these findings can be extrapolated to the IPF population.34 A small randomised controlled trial (RCT) looked at the effects of ambulatory oxygen on IPF patients without resting hypoxaemia but who desaturated during exercise; while oxygen levels were improved, no significant differences in 6MWT distance, level of dyspnoea or leg fatigue were noted.35 The recent British Thoracic Society oxygen therapy guidelines echo the comments from NICE and go further in suggesting that as a ‘good practice point’, the use of ambulatory oxygen may be beneficial as a palliative adjunct in patients with severe dyspnoea but without evidence of hypoxaemia.36 Similar to the NICE recommendations for PR in IPF, RCTs are needed to properly validate this treatment strategy.
Anti-oxidant therapy
The rationale for the use of NAC in IPF was based on evidence that oxidative stress plays a role in the pathogenesis of IPF, through the increased production of reactive oxygen species by cells lining the alveoli, leading to cellular dysfunction and activation of fibrogenic pathways. NAC is a precursor of the endogenous antioxidant glutathione which is found in high concentrations in normal lungs but reduced in patients with IPF.37 The IFIGENIA trial was a RCT multicentre trial assessing the efficacy of treatment with NAC, prednisolone and azathioprine. This triple-therapy approach was shown to slow decline of both VC and DLco compared with azathioprine and prednisolone.38 The study however was criticised for the lack of a placebo arm and high dropout rate of 30%. The PANTHER-IPF study was therefore designed to address this and looked at the efficacy of NAC alone and in combination with prednisolone (at a surprisingly high dose of 40 mg) and azathioprine compared to placebo. The triple-therapy arm was halted following a pre-specified interim analysis showing an increase in mortality, hospitalisations and adverse effects.17 Patients in the placebo and NAC arms of the trial continued and analysis of data at 60 weeks revealed no significant change in the primary endpoint of change in FVC or secondary endpoints including 6MWT, exacerbation frequency and mortality between NAC and placebo.39 The use of NAC for IPF cannot therefore be recommended as a disease-modifying agent, but in a subset of patients in whom chronic cough is problematic, there is anecdotal evidence that use of an anti-oxidant may improve symptom control. Further studies are required to properly evaluate this.
Anti-acid therapy
It is hypothesised that IPF is driven by an abnormal response to trivial but repetitive injury to the alveolar epithelium. One such precipitant may be gastric fluid, resulting from chronic microaspiration in patients with gastro–oesophageal reflux. A recent study using oesophageal manometry to evaluate reflux events found that IPF patients had significantly worse reflux than controls, and pepsin originating from the GI tract has been isolated in bronchoalveolar lavage fluid in patients with acute exacerbations and correlates with a poor prognosis.40 A retrospective analysis of the placebo arm of three IPF RCTs which compared patients on acid suppression with those who were not, found that the use of anti-acid therapy was associated with a slower decline in FVC over time and fewer acute exacerbations.41 Interestingly, research studies have also shown that gastric contents that are not acidic are also capable of causing cellular injury in the lung.40,41 To address the issue of reflux worsening disease, a clinical trial is currently being undertaken in the US comparing decline in FVC in IPF patients who have undergone fundoplication to those who have not (clinical trial number NCT01982968). The recent ATS guidelines conditionally recommend empirical treatment of gastro–oesophageal reflux in IPF patients but acknowledge that good-quality evidence is currently lacking.
Acute exacerbations
Patients with IPF may experience an acute worsening or exacerbation of their disease resulting in increased breathlessness and hospital admission in the majority of cases. Acute exacerbations carry an exceptionally poor prognosis with a three-month mortality of over 50% and a significant step-down in functional status in those who survive. The term ‘acute exacerbation’ has been defined by consensus opinion as: a worsening of dyspnoea over 30 days or less, new airspace changes on HRCT, and exclusion of other causes such as heart failure, pulmonary emboli and infection.42–44 More recently, the necessity of excluding infection has been questioned due to the realisation that it is often not clinically feasible to exclude infection with certainty (bronchoscopy often contraindicated on the basis of low oxygen saturations for example) and increasing evidence that infection may act as an initial trigger for an acute deterioration in the disease process.45 In a subgroup analysis following the STEP-IPF trial, investigators analysed the outcomes of patients with ‘definite acute exacerbations’ (in which infection and alternative causes for decline had been excluded) to patients with ‘suspected acute exacerbations’ who had identical features of acute exacerbations but without the stringent exclusion of associated causes. No differences between the two groups were demonstrated, including quality-of-life measures, 6MWT, lung function parameters or mortality.46 Thus ‘acute exacerbation’ is probably best considered a term that describes a clinical and radiological decline that is likely to be caused by a number of triggers (of which infection is probably the most common), but in a proportion of patients may be a truly idiopathic phenomenon.
At present, relatively little is understood regarding the pathogenesis of acute exacerbation and it is unclear whether these events represent an acceleration of the disease process or a separate pathological event within a predisposed diseased lung.44 The histological picture is that of diffuse alveolar damage which is an acute and non-specific reaction of the lung to a multitude of injurious agents, and is characterised by the presence of hyaline membrane formation and oedema following endothelial and alveolar cell injury.47
Treatment for acute exacerbation is currently very limited and entirely without an evidence base. Antibiotics are invariably prescribed to treat possible infection, regardless of the clinical findings, and in the majority of cases, patients are treated with high-dose prednisolone, usually in the form of pulsed methylprednisolone for three days. The rationale behind its use is the notion that the deterioration in the disease may in part be caused by a superimposed inflammatory component within the scarred lung. While a subset of patients respond well to steroid therapy, in a significant proportion of cases it does not appear to change the course of the decline. Therefore, further research and clinical trials extending to patients with acute exacerbations are essential to improve the outcome of this often fatal complication.
Current and future trials
After the disappointment of multiple previous clinical trials investigating drugs that failed to show any advantage and may have even harmed patients with IPF, the development of two anti-fibrotic drugs that clearly improve the rate of lung function decline represents a major advance in the management of patients with IPF. However, pirfenidone and nintedanib are not the panacea and it is worth noting that the clinical trials found no significant differences in quality-of-life measures. These drugs slow progression, but over time the disease will continue to worsen, albeit at a slower rate. If present, the side effects experienced by patients are usually ‘mild’ or ‘moderate’ but for some patients may outweigh any perceived long-term benefits. Thus, while the development of effective anti-fibrotic therapy provides much-awaited treatment options, there is significant clinical and commercial appetite to develop alternative, possibly more effective agents to be used either alone or in combination with others. It is likely that combination regimes to tackle different parts of the fibrotic pathway will be required, akin to regimes used in tuberculosis and cancer. Future trial design is likely to be influenced by the availability of effective therapies, potentially limiting the feasibility of a placebo arm.
Summary
In summary, a long overdue focus on the robust and multidisciplinary diagnosis and management of IPF, combined with significant breakthroughs in therapeutic strategies, makes this an exciting time in the IPF field. The emphasis on specialist ILD centre involvement being at the centre of IPF management has led to necessary and significant service re-design, and hopefully will improve the historical therapeutic nihilism associated with the diagnosis; the most common reason that anti-fibrotic treatment is not possible is late referral of patients with advanced disease. Early and accurate disease phenotyping and early consideration of anti-fibrotic therapy in order to prevent, rather than to react to, decline provides options to alter the devastating natural history of IPF. Finally, a multidisciplinary and holistic approach is key, with emphasis on specialist nurse-led education, drug counselling, side-effect monitoring and symptom-based measures being central to gold-standard patient management.
- © Royal College of Physicians 2016. All rights reserved.
References








{kind=link}
{kind=link}
Jump to section
- Article
- ABSTRACT
- Background
- Defining the diagnosis of IPF
- Overall management principles
- Approaches to management
- Pharmacological treatment options
- Pirfenidone
- Nintedanib
- Managing side effects of pirfenidone and nintedanib
- Other considerations
- Pulmonary rehabilitation
- Anti-acid therapy
- Current and future trials
- Summary
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.