
ABSTRACT
Descriptions of motor neuron disease (MND) documented more than a century ago remain instantly recognisable to the physician. The muscle weakness, typically with signs of upper and lower motor neuron dysfunction, is uniquely relentless. Over the last 30 years, a wider cerebral pathology has emerged, despite the lack of overt cognitive impairment in the majority of patients. From the initial linkage of a small number of cases to mutations in SOD1, diverse cellular pathways have been implicated in pathogenesis. An increasingly complex clinical heterogeneity has emerged around a significant variability in survival. Defining a cellular signature of aggregated TDP-43 common to nearly all MND and a large proportion of frontotemporal dementia (FTD), has placed MND alongside more traditional cerebral neurodegeneration. With new genetic causes, most notably a hexanucleotide expansion in C9orf72 associated with both MND and FTD, the development of biomarkers against which to test therapeutic candidates is a priority.
Introduction
Once seen, motor neuron disease (MND) is never forgotten. Among the adult neurodegenerative disorders it is one of the most rapidly progressive and physically transforming. Advancing weakness of limb and frequently bulbar musculature leads to increasing dependency in the activities of daily living, with death typically from the complications of chronic respiratory failure. The median survival is just 30 months from the onset of symptoms.1 MND has a reputation, beyond the stereotyped view of neurological disease more widely, for having taken up permanent residence in a therapeutic wasteland. Over the last 30 years, however, advances in the understanding of the molecular pathogenesis of MND in conjunction with the characterisation of a heterogeneous clinical syndrome fundamentally involving the brain, is making a future therapeutic era a realistic aspiration.
Traditionally, MND is defined by the relatively selective loss of upper motor neurons of the corticospinal tract in combination with a variable loss of lower motor neurons of the brainstem nuclei and spinal anterior horn cells. This classical combined syndrome of muscle wasting secondary to lower motor neuron loss (amyotrophy), in conjunction with sclerosis of the lateral corticospinal tracts (and the associated upper motor neuron signs of hyperreflexia and spasticity), was termed amyotrophic lateral sclerosis (ALS) by Jean-Martin Charcot in 1865. However, clinical, neuroimaging, immunohistological and genetic advances over the last 30 years have revealed MND to be a complex syndrome that increasingly defies simple taxonomy.2 Even within ALS, the commonest clinical phenotype of MND, multiple upstream pathways are recognised. The dogma of MND as a neuromuscular rather than primarily cerebral degenerative disorder, and one sparing the higher cognitive functions, persisted for more than 100 years. However, MND is now understood to overlap with frontotemporal dementia (FTD)3 and shares many of the core prion-like protein misfolding, aggregation4 and mislocation processes5 common to Alzheimer’s, Parkinson’s and other neurodegenerative disorders. This is bringing MND to the forefront of what was traditionally dementia research.
An unmistakeable clinical syndrome
Despite the increasing molecular complexity of MND, the core syndrome of ALS is as unmistakeable today as it is in the earliest written descriptions. Frederick Mott (1863–1926), writing in 1895, described in painstaking detail a case of ALS in a 39-year-old dressmaker.6 The contemporary mean age of symptom onset in ALS is around 65 years of age in population-based studies. However, the ALS literature of Mott’s time is strikingly dominated by much younger individuals, and this may be more complicated than simply a factor of increased life expectancy.7
Mott noted in passing that the patient had ‘nervous debility in (the) family’. The basis for Mendelian genetics was not understood at this time. At least 90% of MND patients will not record a family history of the condition (or the related condition FTD). One of the fathers of clinical medicine, William Osler, described the first familial cases of MND in the Farr family of Vermont in 1880, whose later generations provided the first genetic linkage of MND to mutations in superoxide dismutase-1 (SOD1).8 Osler was fascinated by the kind of person who developed a given disease, as much as the nature of the disease itself. In Mott’s case, ‘no previous illness of a serious nature’ was noted. This could be said for many adult-onset serious illnesses, but many contemporary neurologists are struck by the premorbid physical fitness of MND patients. Epidemiological studies continue to debate the issue of whether ALS patients undertake significantly increased premorbid physical activity,9,10 and the confounded observations of lower cardiovascular morbidity11 and body mass index12 prior to diagnosis. Methodological constraints of retrospective, questionnaire-based analysis and untangling association with cause continue to obfuscate. The received wisdom that MND patients have a significantly more agreeable personality type has no scientific support to date and is heavily confounded by the recognition of frontotemporal cognitive and behavioural impairment in at least one third of MND patients, which may be associated with apathy.13
Progressive, typically painless paralysis is at the core of the clinical syndrome of MND (‘3 Ps’). Mott recorded that his patient
had been troubled with a feeling of cold in the right leg, from knee to toes, and a sense of weight in walking. The difficulty increased so that she dragged the right leg, which felt heavy and cold, and it commenced to waste.
Such progressive weakness and wasting of a single limb is a common first manifestation of ALS. Patients easily conflate weakness with sensory impairment (cf Bell’s palsy), and may report a weak limb to be ‘dead’ despite, as Mott noted in his patient, ‘sensation in no way affected’. Pain is a more frequent comorbidity than traditionally considered in MND14 but rarely is it the predominant initially reported symptom.
The site of onset of the first muscle weakness in MND is not entirely random. In 25% of cases it will be bulbar muscle weakness, typically with progressive dysarthria prior to dysphagia. In most of the remaining cases, the initial symptom will be either upper (weak shoulder or hand grip) or lower limb weakness (often foot drop), with a predilection for the dominant side in those with upper limb onset.15 A small proportion of MND patients (<5%) develop initial symptoms related to respiratory failure due to unusually early diaphragm weakness, but more often this occurs in the later stages and is the main driver of mortality.
The spread of symptoms in MND is typically to contiguous body regions,16 with observations from post-mortem examination demonstrating the highest burden of spinal cord pathology at the level of first symptom onset. Mott noted ipsilateral spread in his case:
The right arm began to fail twenty-one days ago; gradually progressive weakness came on, just like the (right) leg, but especially in the thumb and forefingers, and she experienced a difficulty in holding things. Exaggerated knee-jerks present on both sides, and ankle clonus especially marked on the right side.
The majority of MND patients develop clinically demonstrable combined upper and lower motor neurons signs on examination, which currently forms the basis for a firm diagnosis in the setting of progressive weakness. Taxonomies of upper versus lower motor neuron predominance, site of onset, regional spread and speed of progression are all recognised but have significant overlap (Table 1).
The overlapping clinical taxonomies of MND (ALS)
The emergence of the anterior brain
Vision quite good. Hearing good. Nothing definitely abnormal, pulmonary, or cardiac.
These observations in Mott's case speak to the long-held sense that MND is a disorder of selectively vulnerable neuronal populations. One of the most striking aspects of MND is the relative sparing of the oculomotor neurons and those of Oluf’s spinal sphincter nucleus. It is a pivotal, yet relatively unappreciated, observation that these two sets of motor neurons are distinct in not receiving direct cortical projections.17 Undoubtedly, the motor system is dominated by the corticospinal tract and downstream muscle projections. The concept of MND as a corticomotorneuronal (corticofugal, rather than a ‘dying back’ disorder of the neuromuscular junction) disorder was first postulated by Eisen18 and is in keeping with broader evolutionary considerations in relation to the clinical syndrome, linking ‘tools and talk’.19 The heterogeneity of clinical upper and lower motor neuron involvement seems likely to reflect the complexity and variability of divergent cortical projections, converging on downstream lower motor neuron pools.16
Despite some of the earliest published literature noting that patients with MND occasionally displayed cognitive and behavioural disturbance, the dogma that MND spared the intellectual functions persisted until the 1980s. More detailed neuropsychological assessments, supported by early radiotracer brain imaging, identified a deficit of the anterior brain, in particular the executive functions of the frontal and temporal lobes. Over the next 20 years, this was refined to an understanding of MND as a clinicopathological spectrum with FTD.20 Up to 15% of MND patients achieve criteria for FTD, which is typically an early, even pre-motor presenting symptom, and the disease then often follows a more aggressive course. The remaining patients have a more subtle, often sub-clinical syndrome of executive dysfunction or mild behavioural change that is less clearly progressive in comparison to the motor system decline, but is an independent negative prognostic factor.21
The expanding genetic and cellular complexity
Charcot’s ‘debris’, which he noted in the cytoplasm of degenerate neurons, was later identified as a characteristic set of ubiquitinated inclusions; some skein-like, others more granular.22 This was put to one side with the discovery of genetic linkage to a mutation in the SOD1 gene in an ALS family. This led to the first animal model of MND, a transgenic, over-expressing mutant G93A SOD1 mouse with characteristic hind-limb wasting, which until recently has been the mainstay of efforts to screen candidate therapeutics.23 Pathogenic themes of oxidative stress, including mitochondrial dysfunction, grew rapidly and remain a core part of a wider cellular pathological network that encompasses astrocytes and microglial influences on neurodegeneration (Table 2).24 The broadly anti-glutamatergic drug riluzole was shown to extend survival in MND by approximately 10%. Subsequent studies, principally using transcranial magnetic stimulation, have demonstrated cortical hyperexcitability to be a core feature of MND.25 While riluzole remains licensed as a disease-modifying therapy, its effects are undoubtedly modest and not quantifiable at the level of the individual patient. In parallel, an equal if not greater survival benefit has come from the increasing availability of multidisciplinary care, including symptomatic interventions such as gastrostomy to improve nutrition and non-invasive ventilation that has a clear survival effect too.26
Molecular pathogenic themes in the syndrome of MND4,5,24,28
The most important molecular discovery in MND to date was the identification of the ubiquitinated protein common to 98% of cases of MND and crucially, also 50% of FTD, namely the 43 kDa transactive region DNA-binding protein TDP-43.27 This cemented the concept of MND and FTD as a clinicopathological continuum and simultaneously opened up the field of RNA misprocessing as a core element of pathogenesis, supported by the discovery of MND-causing mutations (albeit responsible for <1% of all ALS) in both TARDBP and FUS, united by both being major RNA-splicing and regulatory genes.28
A GGGGCC repeat expansion in C90rf72 was identified in 2011 and found to be associated with up to 10% of all MND in western countries (Fig 1).29,30 Crucially, the gene was also linked to a high proportion of patients with FTD; ‘pure’ ALS, FTD and mixed ALS-FTD cases are seen in the same pedigree, opening up a new frontier in trying to understand selective vulnerability across the neurodegenerative disorders. More than any other to date, the biology of this genetic mutation is revealing novel cellular processes, including nucleocytoplasmic transport deficits that may be at the core of MND and FTD.5,31 Gene silencing oligonucleotide-based strategies are now a realistic proposal.32
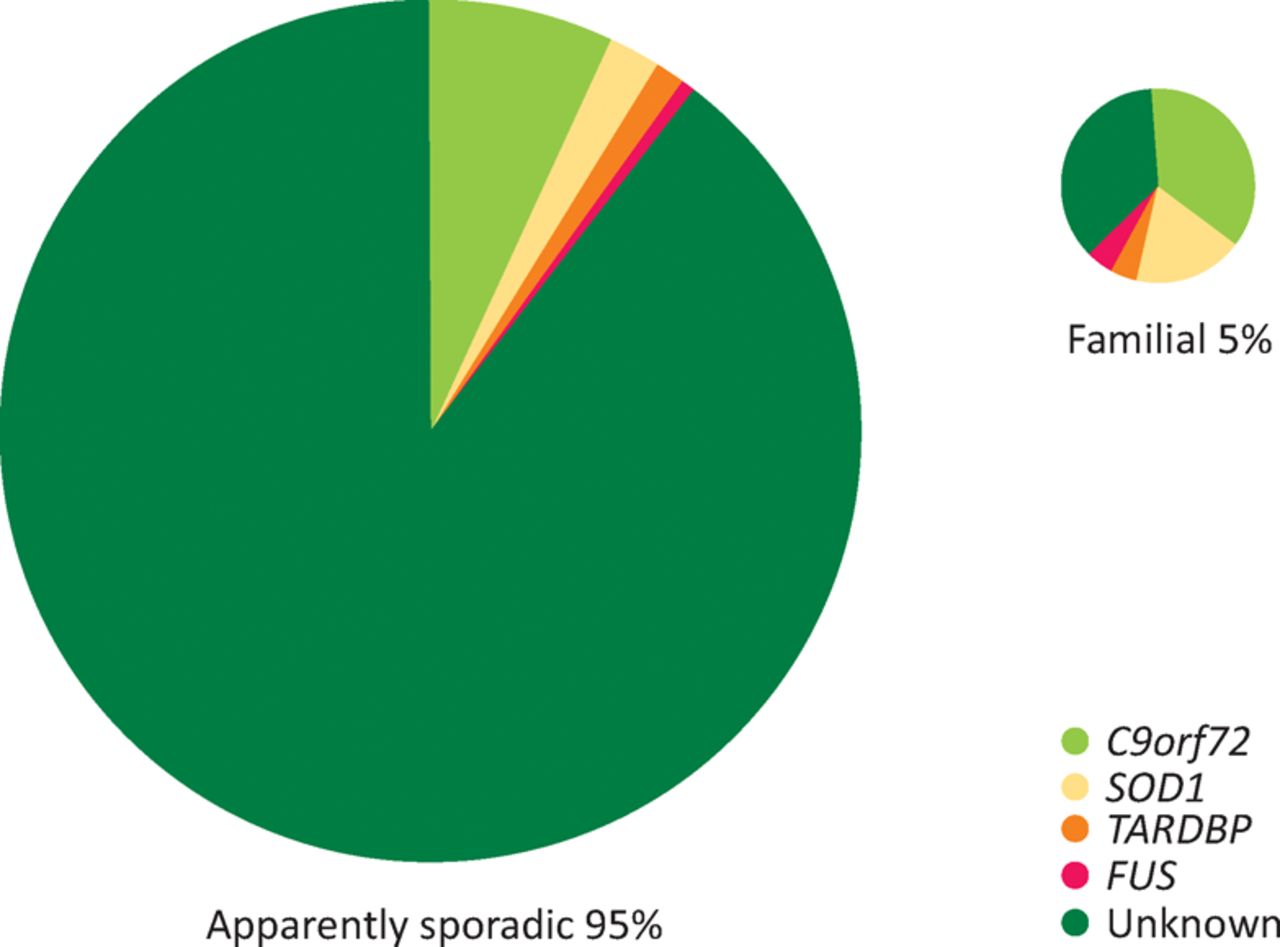
The broad genetic landscape of motor neuron disease (MND). Approximately 5% of all cases report a positive family history in a first-degree relative. Even in those apparently sporadic cases, a significant proportion are positive for an expansion in C9orf72, which is associated with nearly 10% of all cases of MND in the western hemisphere.
Finally, an emerging broader concept is of prion-like spread of pathology through neuronal connections.33 This is based on the common theme of protein misfolding and aggregation across neurodegenerative diseases and the observation of consistent ‘stages’ of pathological involvement in post-mortem brain studies.34
Clinical heterogeneity and the biomarker need
MND is a clinical diagnosis and certainty rises with the demonstration of mixed upper and lower motor neuron signs within body regions (bulbar, arms, legs) that clinically define ALS and are demonstrable in 80% of patients. The insidious onset of MND, linked to a failure of recognition of neurologically mediated symptoms and frequent mis-referral in primary care, all contribute to the average delay of 1 year from the onset of symptoms to firm diagnosis. The role of electromyography is in the detection of occult lower motor neuron involvement (muscle denervation). It has no mandatory diagnostic nor disease monitoring role.35
Although the median survival in MND is 3 years from symptom onset, the second half of the survival curve extends into the second decade. The gold standard for measuring the rate of disease progression is currently a functional disability score, termed the revised ALS Functional Rating Score (ALSFRS-R). The rate of deterioration in an individual patient is generally stable, so that MND is highly individualised despite the variation in site of initial symptom onset. Patterns of regional involvement, such as bulbar-onset to symptoms, tend to be associated with more rapidly-progressive disease but not consistently. Conversely, the ‘flail arm’ pattern of symmetrical proximal upper limb weakness with prominent lower motor neuron signs is uniformly a much slower disease course, typically up to 10 years.36 An objective biomarker of disease activity, more sensitive than the slope of decline of ALSFRS-R to variation in the rate of progression, would be valuable to reduce therapeutic trial heterogeneity and provide faster no-go decisions.37
Rapidly progressive MND is associated with a shorter diagnostic delay as the clinical syndrome is unmistakable in its relentless progression, even to the non-neurologist. The major value of a diagnostic biomarker in MND is therefore in the more slowly-progressing patients who more often have a lower motor neuron-predominant form of MND, which may be confused for an inflammatory neuropathy for example. The detection of occult upper motor neuron involvement in such patients, as well as markers of disease progression, is being explored using advanced magnetic resonance imaging (MRI) and cerebrospinal fluid (CSF) analysis.
Insights from neuroimaging
Early radiotracer single-photon emission computerised tomography (SPECT) and positron emission tomography (PET) activation studies revealed reduced blood flow and metabolism in the anterior brain in MND patients with cognitive impairment.38 Standard MRI of the brain in MND is typically normal, surprisingly without visible atrophy of the motor cortex in most cases and with more widespread frontotemporal atrophy only in those with obvious cognitive impairment. The development of automated segmentation and analysis software applied to high-resolution volumetric MRI scans has been able to demonstrate more subtle but consistent motor and frontotemporal cortical atrophy across a range of MND patients, including those without overt dementia.39 In combination with diffusion tensor imaging of large white matter tracts, a signature of MND has emerged that consistently involves the corticospinal tracts and corpus callosum,40 including in those with no apparent upper motor neuron signs.41 In longitudinal study, more extensive grey matter involvement – including the thalami – becomes apparent.42 Combined studies of structure with function, assessed using blood oxygenation level-dependent (BOLD) MRI in the resting state, indicate motor, pre-motor and medial frontal regions of increased connectivity associated with MND; this is highest in those with faster rates of disease progression.43 At present, the challenge is to translate these MRI findings so that they are applicable to individual patients.
Prospects for a neurochemical biomarker
Neurofilaments are released into the CSF as a result of axonal degeneration associated with a range of neurodegenerative diseases and traumatic brain injury. Although not specific to MND, significantly higher levels are found in MND patients compared with both healthy and a range of relevant disease controls. This is now a leading diagnostic biomarker and further multicentre comparative studies, including in lower motor neuron mimic populations, are underway with the aim of translation to the clinic.44,45 The level of CSF neurofilaments also seems to be linked to the rate of disease progression and in one longitudinal study this level remained fairly stable.46 This has value as a potential outcome measure for therapeutic trials.
The pre-symptomatic landscape
Like all organ systems, the nervous system seems likely to have significant redundancy, so that there is a sub-clinical period of pathology accumulation. Around 10% of all ALS is associated with a hexanucleotide expansion in C9orf72 or pathogenic mutations in SOD1. Asymptomatic carriers of genetic mutations offer a unique window into the earliest changes in MND. CSF neurofilament levels do not appear to rise until just prior to the development of symptoms,47 but there is detectable structural difference prior to symptoms,48 and early clues to subtle changes in brain function.49 This offers the eventual hope of neuroprotective strategies.
Looking ahead
In the last 30 years, MND has been acknowledged as a complex syndrome inextricably linked to the brain through advances in neuropsychological assessment, neuroimaging and genetics. The molecular complexity of the disease has increased exponentially, and is undoubtedly daunting. Akin to cancer, MND is the final common pathway of motor neuron loss that is accessible via multiple upstream pathways. Yet the clinical syndrome remains as striking today as it was to Charcot. More refined non-human MND models based on core biology, and the increasing availability of induced pluripotent stem cell-derived motor neuron cultures, offer the early hope of high-throughput drug screening but less certain potential for cell-based therapy. Neuroimaging and CSF neurochemistry, coupled to deeper clinical phenotyping, are poised to deliver the biomarkers required to translate these to the patients and ignite a therapeutic era in MND.
Conflicts of interest
The author has no conflicts of interest to declare.
- © Royal College of Physicians 2016. All rights reserved.
References








{kind=link}
Jump to section
- Article
- ABSTRACT
- Introduction
- An unmistakeable clinical syndrome
- The emergence of the anterior brain
- The expanding genetic and cellular complexity
- Clinical heterogeneity and the biomarker need
- Insights from neuroimaging
- Prospects for a neurochemical biomarker
- The pre-symptomatic landscape
- Looking ahead
- Conflicts of interest
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.