
ABSTRACT
Monoclonal antibody therapeutics have been approved for over 30 targets and diseases, most commonly cancer. Antibodies have become the new backbone of the pharmaceutical industry, which previously relied on small molecules. Compared with small molecules, monoclonal antibodies (mAbs) have exquisite target selectivity and hence less toxicity as a result of binding other targets. The clinical value of both mAbs and ligand traps has been proven. New applications of mAbs are being tested and mAbs have now been designed to target two (bi-specific, eg TNF-α and IL-17) or more targets simultaneously, augmenting their therapeutic potential. Because of space limitations and the wide ranging scope of this review there are regrettably, but inevitably, omissions and missing citations. We have chosen to highlight the first successes in inflammatory diseases and cancer, but a broader overview of approved mAbs and related molecules can be found in Table 1.
Introduction
There has been a revolution in therapeutics over the past 20 years, due to the introduction of protein therapeutics – often termed ‘biologics’ – into routine, mainstream medicine. Biologics are expensive and like all medicines have risks. Their advantage is exquisite specificity due to greater surface area binding, resulting in decreased ‘off-target’ effects as compared with most small molecule drugs.
New biologic therapies come in several basic forms, either growth factors and cytokines (such as erythropoietin, G-CSF, interferon, enzymes, factors that regulate coagulation) or, more commonly, monoclonal antibodies (mAbs) and related proteins such as ‘traps’ in which cytokine receptors are made soluble and fused with antibody constant regions. The latter group (mAbs and traps) have dramatically advanced the therapy of chronic inflammatory diseases and cancer. In this review, we will focus on mAbs and relatives, describing these therapeutics. The tumour necrosis factor (TNF)-blockers for autoimmune/inflammatory diseases are the most broadly deployed (with multiple products) and have engendered a revolution in therapeutic research/development, along with rather remarkable revenues. This therapeutic revolution is based on the synergy of three scientific disciplines: immunology, molecular biology and protein engineering.
Brief history
The first mAbs were produced by Milstein and Köhler (Cambridge University),1 who won the Nobel Prize in Physiology or Medicine in 1984. mAbs were initially murine proteins and hence were immunogenic in humans and not suited for chronic therapy. This limitation was solved using molecular biology and protein engineering to create more human-like mAbs with little immunogenicity. There are several versions of mAbs and traps in clinical use and trials (Fig 1, Table 1). Initial attempts replaced most murine Fc sequences with human Fc, a process referred to as ‘chimerisation’.2 This involves grafting the murine antigen binding Fab regions onto a human immunoglobulin (IgG) backbone. In ‘humanisation’, the mouse hyper-variable peptide binding loops3 are grafted onto the human IgG backbone. More recent technologies allow for generation of fully human antibodies.4
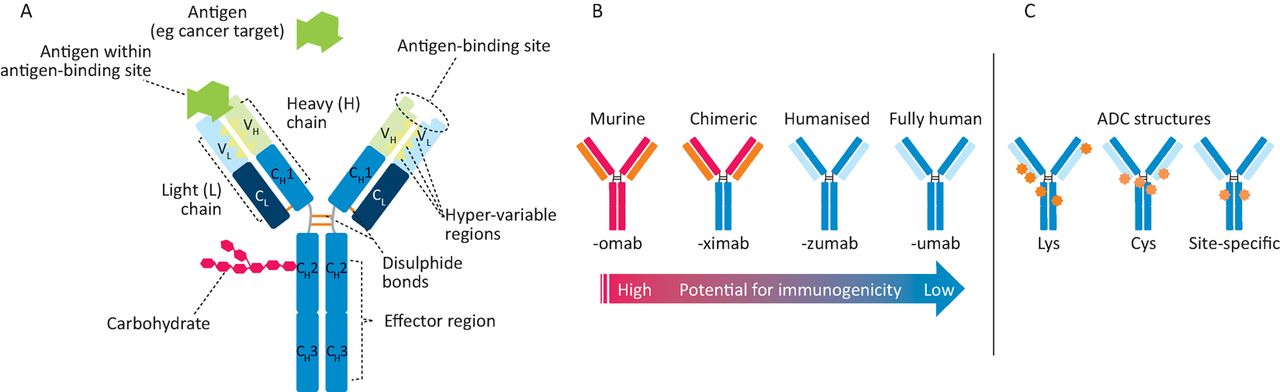
Monoclonal antibodies. A – antibody structure showing light (L) and heavy (H) chains with variable (VL, VH) and constant (CL, CH) regions, which are connected by inter-chain disulphide bonds. Antigen binding (green symbol) occurs at VH/VL domains, while effector functions are mediated via the Fc (CH) portion. B – increasing the amount of human sequences in a murine antibody decreases immunogenic potential. C – on an antibody-drug conjugate (ADC, cytotoxic drugs (orange, red stars) can be linked stochastically to lysine (Lys) residues or on cysteine residues either through reduction of inter-chain disulphides (Cys) or by engineering in cysteines at select conjugation sites (site-specific).
Targets and approvals for monoclonal antibodies and derivatives (asterisks denote approvals in USA only)
Improved therapy for inflammatory/autoimmune disease
The concept of autoimmune diseases, pioneered by Sir Frank McFarlane Burnet5 (Nobel Laureate 1960), was validated by the detection of serum autoantibodies in many diseases in the 1960s. This suggested a role for B-lymphocytes, and human leukocyte antigen (HLA) associations documented from the 1970s indicated the role of T-cells when the peptide presenting function of HLA was understood. However, these advances did not lead to new treatments at the time because of the lack of specific therapeutics.
With the advent of mAbs, the role of T-cells was tested in rheumatoid arthritis patients using lytic antibodies to CD4, without clinical benefit.6 Nor did small trials of several other anti-T-cell antibodies, such as anti-CD7. OKT3, a mouse mAb recognising CD37 given to reduce acute rejection in patients with organ transplants was the first monoclonal antibody approved for use in humans although it was later withdrawn.
The success of anti-TNF therapy
The targeting of a single pro-inflammatory cytokine, TNF, to treat a complex disease in rheumatoid arthritis (RA) where multiple pro-inflammatory cytokines were upregulated was based on work using human disease tissue by Feldmann and Maini8 and Brennan et al.9 They analysed cytokine production from joints and cytokine regulation in cultures of rheumatoid synovium in which the majority of the cells survived, producing the mediators generated in vivo. In these cultures, blocking TNF-α reduced the production of many other inflammatory cytokines (IL-1, IL-6, GM-CSF, IL-8 etc),9 thus defining a ‘TNF-dependent cytokine cascade’. The dramatic clinical success of TNF blockade, demonstrated first in late stage RA then in earlier stage disease, also validated this concept.8 Noteworthy was the fact that tissue (bone and cartilage) damage was controlled. But also striking was the heterogeneity of the clinical response, with some individuals close to a cure and others virtually unimproved. The reasons for this are not yet clear, despite much work to try to elucidate the reasons. Genetic differences were an obvious possibility although never established, and recent clinical data demonstrating that non-responders may respond subsequently to anti-TNF has excluded it. Although anti-TNF in humans is relatively safe,10 more infections in patients occur, eg with intracellular organisms, especially tuberculosis. Many large patient registers have documented the long-term benefits of anti-TNF therapy, reducing some complications and maintaining a favourable benefit/risk ratio.11
Currently, anti-TNF is used in RA, Crohn's disease, ulcerative colitis, psoriasis, psoriatic arthritis, ankylosing spondylitis and juvenile RA; its use is now being explored in other indications (Table 1). Anti-TNF antibodies are the most successful and widely used antibody-based therapeutic. It is noteworthy that if used together with methotrexate early in the course of RA, over 50%12 of patients can be taken off infliximab and remain virtually disease-free, even with reduced dosage of methotrexate,12 and some patients can be taken off all medication.13
In the wake of anti-TNF's success, other mAbs have also proven useful in inflammatory diseases. The antibody to the IL-6 receptor, tocilizumab (Actemra®),14 pioneered by Professor Tadamitsu Kishimoto, has been approved for RA and for juvenile idiopathic arthritis. Currently, other anti-cytokine antibodies to IL-6, GM-CSF and the GM-CSF receptor have been approved (siltuximab (Sylvant®) – anti-IL-6) or are in late stage clinical trials after successful phase II studies.
Rituximab, a chimeric mAb to CD20,15 (an antigen in most B-cells although not plasma cells), was first to treat B-cell-driven cancers such as non-Hodgkin lymphoma. It was pioneered by Jo Edwards for RA and subsequently approved, but was not successful in systemic lupus erythematosus (SLE) trials. Anti-CD52 (alemtuzumab) is a first generation humanised antibody, now used in multiple sclerosis. There are other antibodies approved, eg belimumab (also known as Benlysta) is an anti-BLys mAb approved for SLE,16 ustekinumab (also known as Stelara) is an antibody to the shared p40 subunit of IL-12 and IL-23 approved for psoriasis and psoriatic arthritis and potentially for Crohn's disease,17 and secukinamab (Cosentyx) is an anti-IL17A mAb approved for severe psoriasis and ankylosing spondylitis.18
Receptor IgG fusion proteins (‘ligand traps’) have also been generated. TNF receptor 2-Fc (etanercept, Enbrel®) is effective and has been approved.19 Other ligand traps include rilonacept (Arcalyst – an IL-1 Trap) and the anti-angiogenesis vascular endothelial growth factor trap (aflibercept (Eylea®) for retinopathies and ziv-aflibercept (Zaltrap®) for colorectal cancer) (Table 1). Abatacept (Orencia®) and belatacept (Nolojix®) are CTLA4-Fc fusion proteins that inhibit antigen presentation to T-cells and are effective in RA.
Monoclonal antibodies in cancer therapy
mAb discovery: a case study of trastuzumab (Herceptin)
Modern mAb therapy of solid tumours was initiated by the humanised human epidermal growth factor receptor 2 (HER2) mAb trastuzumab (Herceptin®).20 The rigorous science that laid the foundation for this breakthrough mAb also initiated personalised/biomarker driven drug discovery and treatment in oncology.
Trastuzumab, the first successful monoclonal anti-cancer antibody to be successful against solid tumours, is well tolerated in patients. The pathway leading to TNF-resistance of most tumour cell lines21 was unraveled by collaboration between the Shepard (Genentech) and Schreiber laboratories (Chicago), which revealed that macrophages kill tumour cells largely by secreting TNF.22 They hypothesised that if tumour resistance to macrophages could be reversed, the tumour would become sensitive to killing by host defence. Macrophage (or TNF)-resistant tumour cells implanted into syngeneic mice formed aggressive tumours, while their TNF-sensitive parental cells regressed.21
The mechanism of TNF-resistance of tumours needed to be widespread since most tumour cell lines were resistant. Sporn and Todaro's autocrine growth factor hypothesis23 of malignant transformation involving autocrine stimulation by transforming growth factors seemed plausible. Various growth factors were combined with TNF on TNF-sensitive tumour cell lines and growth factors that activated receptor tyrosine kinases converted TNF-sensitive tumour cells to TNF-resistant cells.24 Host defence was completely subverted and the growth inhibitor (TNF) even became a growth factor.25
Slamon et al26,27 and Zhou et al28 reported that overexpression of the receptor tyrosine kinase HER2 in breast and ovarian cancers was associated with more aggressive disease contemporaneously with Shepard's demonstration that overexpression of HER2 induced resistance to TNF29 and that mAbs to HER2 inhibited growth of HER2-overexpressing tumours in vitro and in vivo.30–32 The lead antibody, MuMAb4D5 – the murine partent of trastuzumab,24was ineffective on normal cells or tumour cells not overexpressing HER2.26,33 Anti-HER2 also validated the importance of stability of the target itself as HER2 is indispensable in driving disease in subsets of breast, gastric and ovarian cancers. These findings laid the foundation for the success of trastuzumab and subsequently for pertuzumab,34 which binds a separate epitope on HER2, and then trastuzumab emtansine, an antibody drug conjugate.35 The clinical success of trastuzumab required the co-development of two novel companion diagnostic approaches: immunohistochemistry and fluorescence in situ hybridisation for selecting patients most likely to respond.36 This was a new idea at the time, but is now a routine part of most cancer drug discovery and therapeutic programmes. Finally, this mAb showed that antibody therapy was feasible in solid tumours, overcoming problems such as high internal tumour pressure, which causes vascular collapse and reverse flow of fluid in solid tumours.37
MuMAb4D5 was humanised using a consensus human variable domain sequence from the Kabat database, together with human immunoglobulin constant domain (IgG1, kappa) sequences.38 This resulted in the synthesis of a recombinant antibody, non-existent in nature, with almost 95% human sequence, reducing concerns related to immunogenicity. The humanised antibody has three mechanisms of action dependent upon upregulated HER2:
growth inhibition
antibody-dependent cell-mediated cytotoxicity (ADCC)
induction of TNF sensitivity.20,30
The strategy employed in the development of trastuzumab is now widely employed in cancer drug discovery (Fig 2). Very recently in clinical trials, a combination of trastuzumab and another anti-HER2 mAb (pertuzumab30) generated a complete pathological response in approximately 50% of HER2-positive patients,39 which is a previously unheard of therapeutic advance in the treatment of breast cancer. The HER2 programme proved that mAbs have a place in therapy of solid tumours and that tyrosine kinase oncogenes were viable targets in cancer.
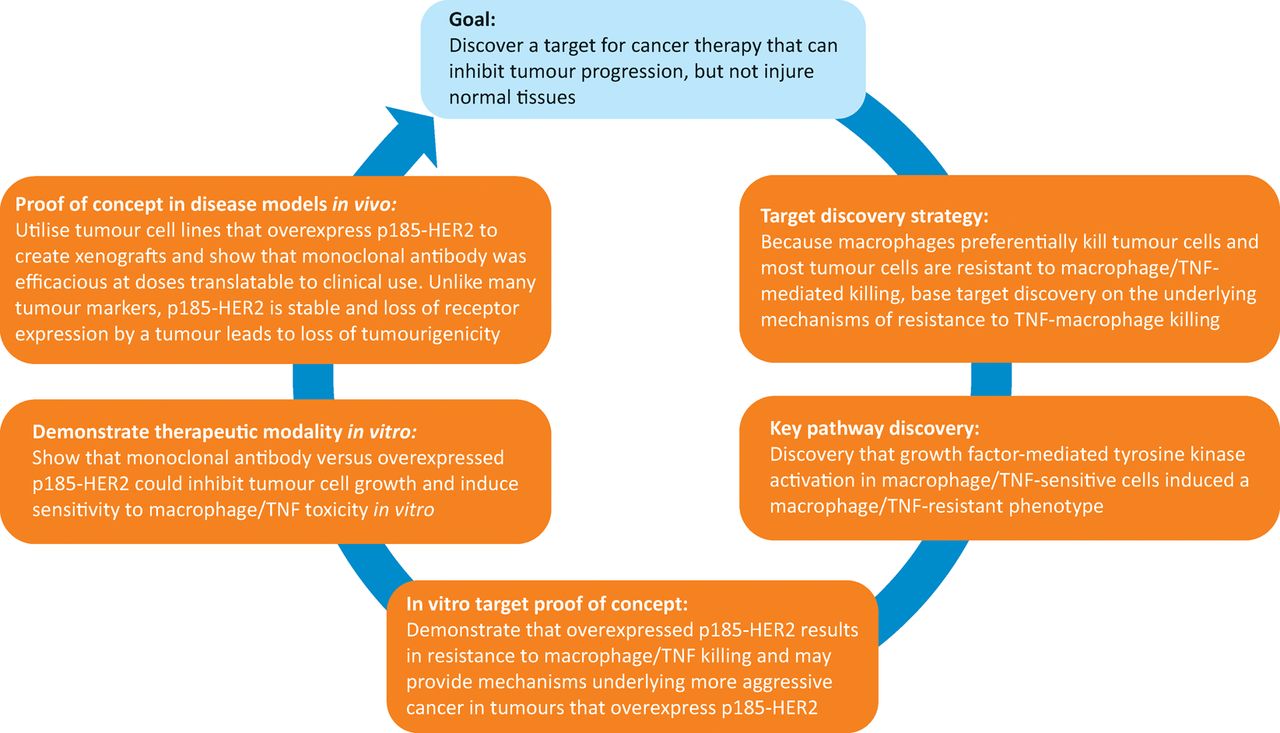
Strategy and key steps for development of trastuzumab, from target discovery to in vitro and in vivo proof of concept studies. HER2 = human epidermal growth factor receptor 2; TNF = tumour necrosis factor
Not all successful mAb therapeutics followed this pathway of discovery. The first approved mAbs targeted membrane proteins shared between haematologic malignancies and their precursor immune cell subsets (eg alemtuzumab (Campath®) – an anti-CD52, rituximab (Rituxan®) – an anti-CD20). These mAbs ablate both normal and cancer cells. Alemtuzumab was the first humanised mAb therapeutic, depleting lymphocytes, monocytes and dendritic cells, useful in diseases such as chronic lymphocytic leukemia. Similarly, rituximab depletes CD20-expressing B-cells. Resistance to these mAb therapeutics occurs with the loss of the target as neither CD52 nor CD20 is essential for malignant growth. These mAbs have toxicity issues due to massive depletion of immune cells, potentially causing ‘cytokine release syndrome’ or ‘tumour lysis syndrome’ – a ‘cytokine storm’ resulting from aberrant immune activation attributable to cellular debris activating ‘danger receptors’. Despite these issues, both rituximab and alemtuzumab are successful drugs and often lead to disease regression.
More recently, Jim Allison and other cancer researchers discovered the utility of mAbs targeting ‘immune checkpoints’. By inhibiting these pathways (eg programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1) or cytotoxic T-cell lymphocyte associated protein-4), tumour-associated immune suppression can be reversed with clinically effective immune cell attack on tumours in about 20% cases. Some patients have had dramatic improvements, notably in metastatic melanoma, and there is no doubt that this is a major step forward. Not unexpectedly there are immune-mediated diseases in a large fraction (about 30%) of treated patients, but this field of mAb therapy is growing very rapidly. CTLA-4 expressed on regulatory T-cells exerts immune suppressive activity. Anti-CTLA-4 prevents CTLA-4 from inhibiting immunity.40 PD-1 is present on ‘exhausted’ T-cells; anti-PD-1 reactivates them. The first approvals of this new class of anti-cancer mAbs were in melanoma with ipilimumab (anti-CTLA-4, Yervoy®) and pembrolizumab (anti-PD1, Keytruda®) in 2015, followed by atezolizumab (anti-PD-L1, Tecentriq®) for non-small cell lung cancer and bladder cancer. Checkpoint inhibitor mAbs, such as anti-OX40, can also be agonistic.40
Other antibody-related approaches and mechanisms
Therapeutic antibodies: structural marvels that mediate multiple functional effects
Antibodies have evolved over the past 400 million years to provide broad ranging host defence in all vertebrates. Thus, it is not surprising that engineered antibodies have emerged as one of the main sources of new medicines. Antibodies evolved to possess several important functional properties exploited by recombinant mAb therapeutics, such as exquisite specificity and affinity toward their antigen targets, recruiting immune system effector components and a long serum half-life.
Antibodies possess a unique tertiary domain structure, folding into a complex quaternary structure. Human serum immunoglobulin is approximately 80% IgG, as are most approved therapeutic antibodies. These complex proteins (approximately 150 kDa) comprise four polypeptide chains – 2 heavy and 2 light chains – with disulphide bonds providing significant rigidity. The heavy chain consists of a variable domain (VH) and three constant domains (CH1, CH2 and CH3). The light chain consists of a variable domain (VL) and a constant domain (CL) (Fig 1A). IgG antibodies are bivalent and have post-translational modifications. Glycosylation in the Fc domain also helps to provide stability and modulates properties such as binding to Fc receptors.41
The antibody/antigen interaction
Several technologies are used to identify novel antibodies: hybridomas, genetically modified mice with human immunoglobulin sequences and phage or yeast display. B-cells from a mouse injected with an antigen are fused with immortalised B-cell myeloma cells to make hybridomas. To improve their efficacy in patients, protein engineers created chimeric antibodies containing human constant domains and murine variable domains, which reduce immunogenicity and activate effector functions (Fig 1). Approved chimeric antibodies include Remicade® (infliximab), Erbitux® (cetuximab) and Rituxan® (rituximab). As an example of a ‘humanised’ antibody Herceptin® (trastuzumab) was isolated from a murine hybridoma and then ‘humanised’ where all but the binding site to the HER2 antigen was changed to a human sequence (Fig 1). Genetically engineered mice have been developed that can make fully human antibodies, eg Vectibix® (panitumumab) – anti-epidermal growth factor receptor. Another important approach employs synthetic (human) antibody libraries that are displayed on the surface of yeast or phage, which is useful for less immunogenic targets. There are advantages and limitations to each of these methods; therefore, antibody discovery groups will attempt multiple approaches in parallel against a particular target.
Antibody effector function
Immunoglobulins can stimulate immune defence mechanisms through ADCC, antibody-dependent cellular phagocytosis and complement-dependent cytotoxicity. During ADCC, immune cells – usually natural killer (NK) cells – lyse a target cell with an IgG bound to a cell surface target. CD16 Fc receptors expressed on NK cells bind to the Fc region of the IgG, forming a lytic synapse between the NK cell and the target cell, destroying the target cell. During complement-dependent cytotoxicity, complement is recruited to the IgG-bound surface pathogen through C1q binding to the antibody Fc domain, triggering proteolytic events, yielding a membrane attack complex that disrupts cellular membranes with cell death. Many therapeutic antibodies use ADCC as a key mechanism of action, eg trastuzumab and rituximab. Methods have been developed for increasing or decreasing antibody effector function, the number of epitope binding sites or attaching cytotoxic drugs. Some of these strategies are shown in Fig 1.
Antibody pharmacokinetics
Antibodies have a long serum half-life, typically lasting 2–4 weeks in circulation. Antibody binding the Fc domain of the neonatal Fc receptor, FcRn, recycles the antibody, protecting it from other clearance mechanisms. This long half-life with less frequent dosing is often more favourable for patients when extended drug function is needed. Antibody dosing is typically intravenous or subcutaneous. Oral administration is not feasible because of rapid degradation of the antibody in the gut. In addition, therapeutic antibodies cannot pass in appreciable quantities through the intact blood-brain barrier.
Antibody-drug conjugates: using antibodies for tumour-selective delivery of cytotoxics
The therapeutic use of antibody-drug conjugates (ADCs) is based on antibody-mediated tumour-selective delivery of potent cytotoxic compounds. This relies on antibody-induced receptor internalisation, followed by trafficking of the ADC to the lysosomes where the cytotoxic drug is released from the ADC. The free drug (inside the target cell) initiates its anti-tumour activity. Stability of the linker that joins the antibody and cytotoxic agent is key to both efficacy and safety. The linker must be stable in the circulation to prevent premature release of the cytotoxic agent, yet allow release of the cytotoxic drug within the tumour cell. The most common linkers are cleavable linkers such as peptides, cleaved by proteases, or disulphide linkers, which undergo reduction to release the drug within the lysosome. ADCs can also utilise a stable or non-cleavable linker, the free drug is released in the lysosome after lysosomal degradation of the entire ADC molecule.
Despite decades of intense research and development, only three ADCs have gained marketing approval. The first, gemtuzumab ozogamicin (Mylotarg®), was approved then taken off the market. Significant advances were subsequently made in ADC linker design and conjugation technologies, yielding two additional approved ADCs, one for CD30-positive Hodgkin lymphoma and anaplastic large cell lymphoma (brentuximab vedotin (Adcentris®), and one for metastatic breast tumours overexpressing HER2/neu trastuzumab emtansine (Kadcyla®). The anti-CD30 antibody component of brentuximab vedotin is linked to the tubulin polymerisation inhibitor mono-methyl auristatin E through a protease cleavable linker, valine-citrulline. Trastuzumab emtansine is comprised of trastuzumab covalently linked to the maytansinoid derivative, DM1, through a stable, non-cleavable linker. Current research suggests that more efficacious and better tolerated ADCs will be developed, which will provide greater clinical benefit to cancer patients.
Emerging antibody technologies
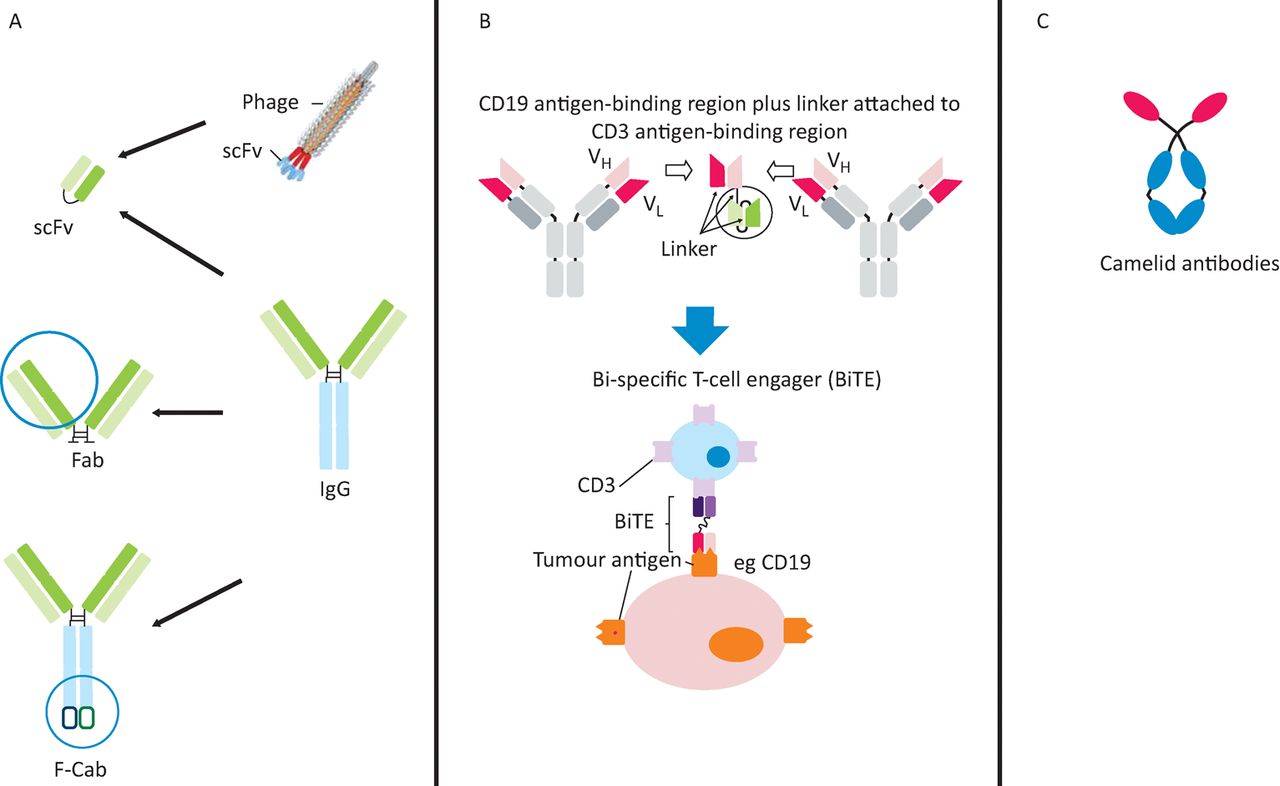
The Fab fragment (Fig 3A) used in ranibizumab (Lucentis)42 can be manufactured in Escherichia coli. The short half-life of Fabs can be remedied by polyethylene glycol used in certilizumab pegol, Cimzia®.43

Emerging antibody technologies. A – derivatives of classical mAbs: scFv, Fab, and F-Cab antibody formats; B – engineered bi-specific mAbs: structure and mechanism of a bi-specific T-cell engaging (BiTE) antibody; C – novel mAb frameworks: unique structure of camelid antibodies. mAb = monoclonal antibody
Until recently, it has been difficult to synthesise bi-specific mAbs. Now various types are possible. Bi-specific T-cell engaging (BiTE) antibodies consist only of the variable regions from distinct antibodies (Fig 3B) and are the format of the first approved bi-specific antibody, catumaxomab (Removab®), for malignant ascites.
Antibody-based cellular immunotherapies
A new class of antibody-based therapeutics called ‘chimeric antigen receptor T-cells’ are emerging in cancer. An antibody-like antigen binding region is fused with signalling components of the T-cell receptor and the chimeric gene is inserted into the patient's own T-cells. When the T-cells recognise their target on tumour cells, the T-cell cytotoxic response is activated, lysing the cancer cells.43 Using CTL019, a chimeric antigen receptor T-cell engineered to target cancer cells that express the CD19 protein present on acute lymphoblastic leukemia cells, 19 of 22 paediatric patients with lethal acute lymphoblastic leukaemia experienced remission.44 However, this treatment upregulates inflammatory cytokines, with consequent toxicity.
Conclusions
mAb therapeutics have been approved for 33 targets in more than 37 distinct diseases (Table 1), most commonly for cancer (27 approvals). The clinical value of both mAbs and ligand traps has been proven. The most targeted pathway is the TNF pathway, now employed to treat nine different indications and likely to expand as TNF is found to be a driver of additional diseases and conditions, including postoperative cognitive decline45 and fibrosis.46 New applications of mAbs are being tested, with approvals in bone metabolism47 and hypercholesterolemia.48
Antibodies have become the new backbone of the pharmaceutical industry, which previously relied on huge libraries of small molecules. Compared with small molecules, mAbs have an exquisite target selectivity and, therefore, less toxicity attributable to binding to other targets. mAbs have now been designed to target two or more targets simultaneously, augmenting the therapeutic potential. We are only just at the beginning of the mAb therapeutic era.
Conflicts of interest
GLP is an employee of Genentech and owns Roche stock. CT and HMS are employees of Halozyme and own Halozyme stock. MF is one of the inventors of anti-TNF therapy and receives royalties on combination therapy.
Author contributions
All authors wrote part of the review, read all of it and approved the final version.
- © Royal College of Physicians 2017. All rights reserved.
References








{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- ioSearch: a tool for searching disease-associated interacting omics; application on breast cancer data
- Proof-of-concept clinical trial of etokimab shows a key role for IL-33 in atopic dermatitis pathogenesis
- Single-domain antibodies represent novel alternatives to monoclonal antibodies as targeting agents against the human papillomavirus 16 E6 protein
- Targeting limited NHS resources to affect optimal care