
Abstract
This article provides an overview of selected genetic skin conditions where multiple inherited cutaneous tumours are a central feature. Skin tumours that arise from skin structures such as hair, sweat glands and sebaceous glands are called skin appendage tumours. These tumours are uncommon, but can have important implications for patient care. Certain appendageal tumours, particularly when multiple lesions are seen, may indicate an underlying genetic condition. These tumours may not display clinical features that allow a secure diagnosis to be made, necessitating biopsy and dermatopathological assessment. Coupled with robust clinical assessment, biopsy findings can guide genetic testing as, increasingly, the causative genes are known for these conditions. Here we review illustrative examples of appendageal tumours and relevant advances made in genetic discovery, and suggest when referral to a geneticist may need to be considered.
Key points
Multiple skin tumours may suggest an underlying inherited genetic condition
A clinical history, family history and full skin examination should be carried out
Skin biopsy and dermatopathological assessment are often necessary to correctly diagnose skin appendageal tumours
Genetic testing should be guided by the clinical phenotype
Genetic counselling and cancer screening may be necessary in affected relatives
Introduction
The skin’s tumour repertoire is diverse, with tumours noted to arise from a range of normal structures. Skin appendageal tumours are derived from skin structures such as hair, sweat glands and sebaceous glands (termed ‘appendages’). These tumours may not display clinical features that allow a secure diagnosis to be made and are typically labelled following biopsy and dermatopathological assessment. When multiple tumours are seen, a rare genetic cause may be the explanation and a cancer predisposition in other organs may exist. Numerous disease-causing genes have now been identified, which allow for genetic counselling should such a syndrome be suspected. Here, we provide an overview of selected inherited appendageal tumours that are seen in tumour predisposition syndromes (Table 1).
Inherited cutanoues tumours and associated systemic features and cancers
Birt-Hogg-Dubé syndrome
Birt-Hogg-Dubé syndrome (BHDS) is characterised by the occurrence of cutaneous lesions on the face, neck and upper trunk.1,2 These lesions are fibrofolliculomas, trichodiscomas and acrochordons. Patients are also susceptible to the development of renal cell carcinoma, lung cysts and pneumothoraces.3
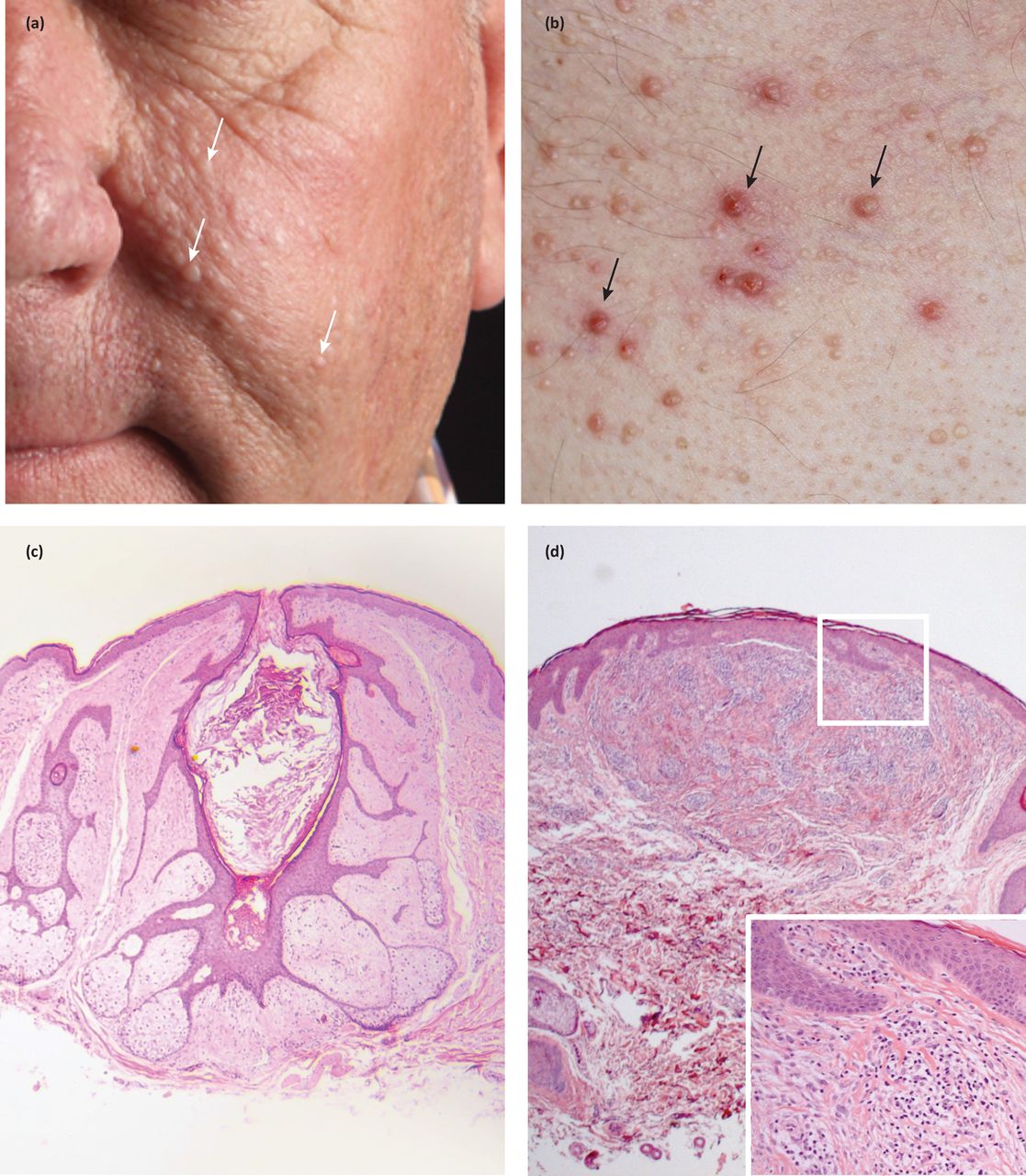
Fibrofolliculomas and trichodiscomas clinically present as skin/yellow-white coloured dome shaped papules 2–4 mm in diameter (Fig 1a and Fig 1b).4 These lesions usually develop in the third or fourth decade.4 In the case of fibrofolliculoma, hair specific differentiation is seen, whereas in the case of trichodiscoma, differentiation is to the mesodermal component of the hair disc (Fig 1c and Fig 1d) .4 Acrochordons are skin tags that were initially described as part of the BHDS; however, they are common in the general population and therefore are not diagnostic of the syndrome.4
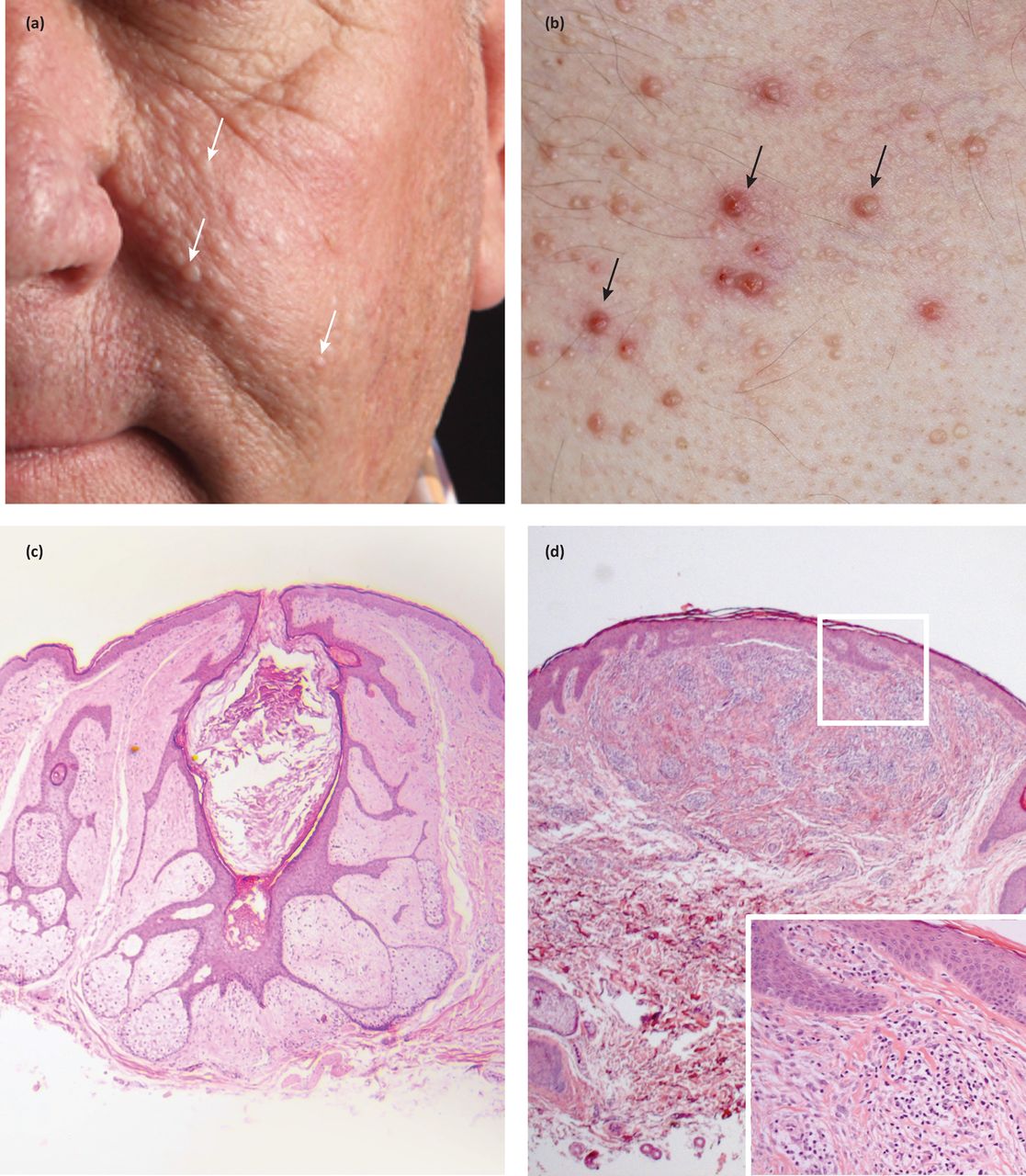
Facial and truncal papules in a patient with a germline FLCN mutation. Facial lesions are seen on the cheeks and nose (a), and are pale or skin coloured papules. Lesions on the chest (b) can be yellow or skin coloured, are sometimes itchy and are consequently excoriated (black arrows). Two tumour types are seen on histology: (c) fibrofolliculomas and (d) trichodiscomas (inflammatory cell infiltrate magnified in inset).
BHDS is associated with a sevenfold increase in risk of developing renal cell carcinoma (RCC). The most frequent type is chromophobe/oncocytic hybrid RCC, found in 50% of cases in a study of 130 tumours across 30 patients with BHDS. The syndrome is also associated with lung cysts in 80% of affected patients, with an associated fiftyfold increase in the risk of pneumothoraces compared with unaffected siblings.1 It is important to note that patients with recurrent pneumothoraces and also patients with a family history of recurrent pneumothoraces should be screened for BHDS. Because of the relatively low incidence of renal cancer and the relatively subtle cutaneous signs, this diagnosis may be overlooked.5
Birt-Hogg-Dubé syndrome is an autosomal dominant inherited genodermatosis attributable to a loss of function mutation in the FLCN gene, which encodes folliculin, on chromosome 17p11.2.1 Folliculin regulates the mTOR intracellular signalling pathway, a pathway also involved in the tuberous sclerosis complex, which shares some phenotypical features in common with BHDS.3 The majority of patients with BHDS will have a detectable mutation in the FLCN gene. Diagnostic FLCN gene analysis is available. Identification of a mutation in this gene not only provides molecular confirmation of the diagnosis but also enables more accurate cascade testing of the patient’s family. Mutation carriers can be offered clinical advice and surveillance for renal tumours, which would typically involve baseline magnetic resonance imaging (MRI) followed by annual renal ultrasound and/or periodic MRI.
Familial leiomyomas
Historically, hereditary leiomyomatosis was clinically associated with two separate but allelic conditions: multiple cutaneous and uterine leiomyomas (MCUL - OMIM 150800) and hereditary leiomyomatosis and renal cell cancer (HLRCC - OMIM 605839). These conditions are now recognised as the same syndrome with phenotypic variance.6 MCUL is characterised by the development of benign cutaneous smooth muscle tumours, uterine fibromas and infrequent uterine leiomyosarcomas. Cutaneous leiomyomas are red/brown skin coloured papules usually 0.2–2 cm in diameter and can be associated with pseudo-Darier sign (transient elevation) on rubbing (Fig 2a and Fig 2b).7 They generally present between the 2nd and 4th decade on the trunk and limbs as firm grouped papules or nodules, and can be tender both in response to touch or reduction in temperature. Cutaneous leiomyomas are present in 76% of patients by the age of 25 years; notably, 40% have mild cutaneous involvement with five or fewer lesions.6 Rapid, progressive growth of a lesion (an infrequent event) should raise suspicion of cutaneous leiomyosarcoma and prompt referral for excision.8

Painful inherited cutaneous tumours. Cutaneous leiomyomas are seen in fumarate hydratase mutation carriers (a, b) and can present as isolated papules but also frequently as clusters in a patient. (c) Cutaneous spiradenomas are typically blue, painful tumours and are a feature of CYLD cutaneous syndrome responsible for familial cylindromatosis (black arrows).
Uterine leiomyomas in affected patients are larger, more numerous and occur earlier than those seen in the general population and are present in almost all female patients by the mean age of diagnosis of 30 years.6 RCC is less common, affecting 20%, and generally occurs in younger patients, with a mean age of diagnosis of 46 years. RCCs are usually highly aggressive, solitary, unilateral and disseminate rapidly. The most frequent histological subtype associated is type 2 papillary RCC.6 Both MCUL and HLRCC are autosomal dominant conditions associated with a heterozygous mutation in the gene encoding fumarate hydratase (FH) at chromosome 1q42.3-a43.9,10FH is thought to act as a tumour suppressor and encodes the fumarate hydratase enzyme, which catalyses the conversion of fumarate to malate in the Krebs cycle.9
The majority of patients with HLRCC will have a detectable mutation in FH. Diagnostic FH sequencing is available. Identification of a mutation in this gene not only provides molecular confirmation of the diagnosis but also enables more accurate cascade testing of the patient’s family. Mutation carriers can be offered clinical advice and surveillance for renal tumours. Surveillance typically employs renal MRI or contrast computerised tomography (CT); there are concerns that renal ultrasound may not be sufficiently sensitive in this context as a surveillance tool although it is useful in monitoring cystic lesions. There are also theoretical concerns about exposing patients with germline mutations in cancer genes to serial CT scans.
Cowden syndrome
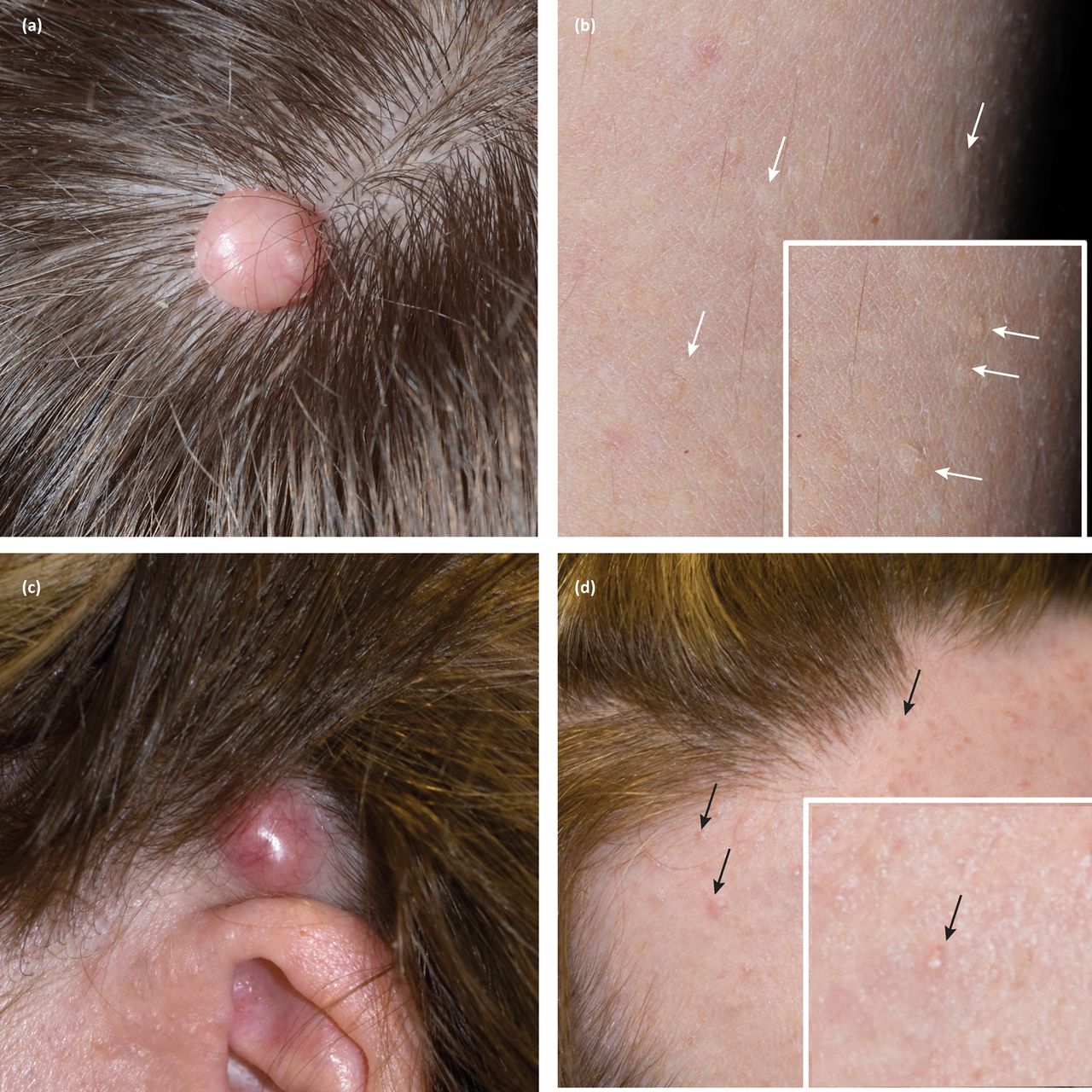
The prevalence of Cowden syndrome (CS) has been estimated at between 1/200,000 and 1/250,000.11 CS is associated with hamartomatous overgrowth of tissues of ectodermal, mesodermal and endodermal origin. Cutaneous features in CS include trichilemmomas (Fig 3b), which clinically present as hyperkeratotic papules typically located at nasolabial folds, nose, ears and upper lip.4 These are benign appendageal tumours with hair follicle differentiation.4 Other features include acral keratosis and verrucous lesions, which are reported in 65–93% of patients.12 Angiolipomas are noted and manifest as palpable subcutaneous nodules that may be tender. Sclerosing collagenomas are uncommon and may be seen in the scalp (Fig 3a). Mucocutaneous features are present in 99% of patients and generally present in the second decade.4 Oral mucosal papillomas and fibromas are reportedly seen in 83–86% of cases; clinically these present as small 1–3 mm papules with smooth surface, primarily found on the gingival, labial and palatal areas. CS can also less frequently be associated with lipomas.12
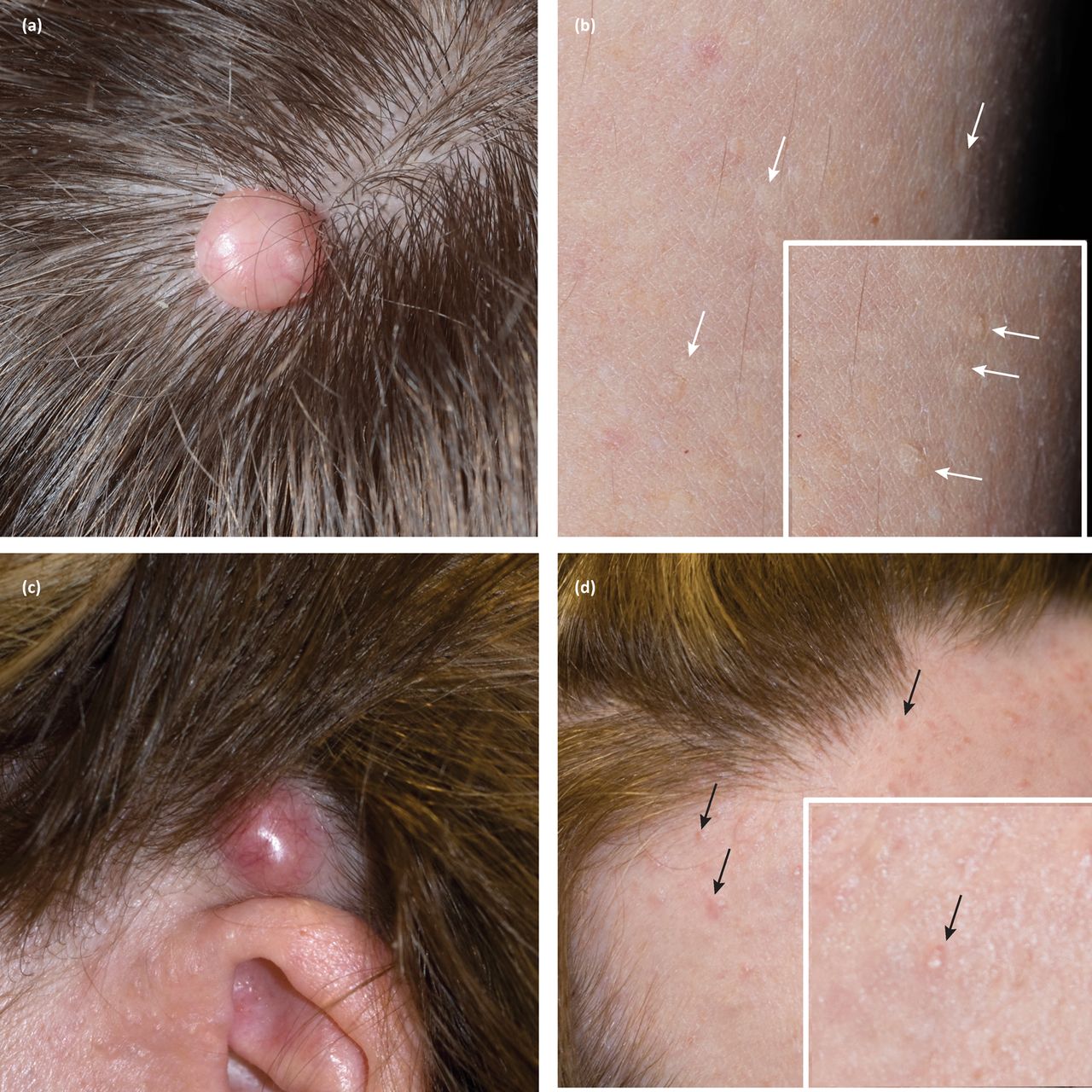
Cutaneous tumours in patients with germline mutations in PTEN and CYLD, with clinical labels of Cowden syndrome and CYLD cutaneous syndrome, respectively. Note the clinical similarity of (a) sclerosing collagenoma and (c) cylindroma, as well as the subtle features of (b) trichilemommas on the limbs (white arrows) and (d) early cylindromas on the forehead (black arrows).
CS is associated with a predisposition for developing both benign and malignant internal tumours. The reported risk for developing breast cancer is two to four times higher than that of the background population, with an average age of diagnosis of 38–46 years; male cases of breast cancer are recognised.11 Thyroid malignancy is also reported (follicular or papillary type) with an increased risk of 3–10% compared with the background population risk of less than 1%. The risk of developing endometrial carcinoma is 5–10% compared with the lifetime risk of 2.5% for the general population.11
Benign breast disease is common in CS with fibrocystic disease reported in 76%. Polyposis of the gastrointestinal tract, primarily hamartomatous, is reported in 40% and benign thyroid disease is reported to affect 50–70% of patients with CS. Patients with CS are reported to be at higher risk of developing haemangiomas with a risk of 22–35% compared with a background population risk of 5–10%.11
CS is an autosomal dominant multisystem disease with incomplete penetrance and variable expressivity. In 80% of patients there is an identifiable germline mutation in the PTEN tumour suppressor gene on chromosome 10. PTEN mutations have been identified in a spectrum of autosomal dominant disorders, including Cowden’s disease, Lhermitte-Duclos disease, Bannayan-Zonana syndrome and ‘PTEN harmartoma tumour syndrome’, which share similar pathological features – including the development of multiple benign tumours, macrocephaly and an increased susceptibility to a range of cancers. Consensus diagnostic criteria have been developed.11 Diagnostic PTEN testing is available and allows diagnostic confirmation and cascade genetic testing. Management comprises monitoring for signs of associated malignant disease, including periodic breast, thyroid and colorectal surveillance. Prophylactic thyroidectomy and mastectomy should be considered on a case by case basis.
Muir-Torre syndrome
Muir-Torre syndrome (MTS) is associated with characteristic cutaneous features, including the development of sebaceous gland neoplasms and keratoacanthomas.13 Sebaceous gland neoplasms are rare, benign tumours that present as either yellow papules or nodules. They are distinct from sebaceous hyperplasia, a relatively common cause of yellow, facial papules. In sporadic cases these tend to be located on the scalp, face and eyelids. In MTS these can frequently be seen on the face, but can also develop on the body.14 MTS has a strong association with colorectal, uterine and ovarian adenocarcinoma; other gastrointestinal malignancies and transitional cancers of the renal tract can also occur.15 Diagnostic criteria include the presence of at least one sebaceous neoplasm together with an internal malignancy or the presence of multiple keratoacanthomas and an internal malignancy in a patient with a family history of MTS.13 Approximately half of patients with MTS will develop two or more visceral malignancies.
MTS is an autosomal dominant genodermatosis, which was initially described independently by Muir in 1967 and Torre in 1968. It is usually a result of a germline mutation in the DNA mismatch repair (MMR) genes and is therefore a subtype of Lynch syndrome (also known as hereditary non-polyposis colorectal cancer). A molecular study of sebaceous adenomas and carcinomas identified Lynch syndrome in 14% of cases.16 Over 90% of cases are found to have mutations in the MSH2 gene. MLH1 accounts for less than 10%. MSH6 has been identified in some MSH2-negative cases.13 In MTS, a germline mutation in an MMR gene in combination with a further somatic mutation affecting the remaining normal allele leads to loss of the MMR protein in a cell. DNA replication errors in a range of cell cycle control genes ensue, leading to the development of both cutaneous and visceral tumours.13 Laboratory diagnosis of MTS is made in tumour samples that demonstrate a genetic feature called ‘microsatellite instability’ or, alternatively, loss of expression of MMR proteins on immunohistochemical analysis. Diagnostic sequencing of the four main MMR genes is available, allowing accurate diagnosis and family cascade testing. This should be offered in accordance with published clinical guidelines.16
These syndromes illustrate the necessity for robust clinical and pathological phenotyping coupled with genetic analysis in patients with multiple inherited tumours. It is important to note that ‘gene sequencing’ is different from ‘genetic testing’. Sequence analysis will detect the majority of mutations; however, a significant number (usually in the region of 5% or so) will be overlooked or missed by sequencing alone. For those patients in which a mutation is not detected by gene sequencing, the genetic test should include screening for whole gene deletions and exonic deletions and duplications. In practice, both approaches are usually combined in a single genetic test. Extended genetic testing might be required in some patients in whom a clinical diagnosis appears secure: for example, sequencing of the PTEN promoter region may be considered when CS is clinically suspected in patients with a negative sequencing result. Similarly, if genetic test results are negative in a patient with MTS, it might be appropriate to consider multiple gene analysis in the form of a ‘gene panel’. It is also evident that patients with cancer predisposition syndromes, such as those described above, often have complex healthcare needs and multidisciplinary specialist input is usually required for the patient and family.
Conflicts of interest
The authors have no conflicts of interest to declare.
Acknowledgements
Neil Rajan is Wellcome intermediate clinical fellow.
- © Royal College of Physicians 2017. All rights reserved.
References








{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.