
ABSTRACT
The autoimmune cytopenias are a group of disorders resulting primarily from autoantibody-mediated destruction of blood cells, with variable clinical sequelae depending on the severity and lineage affected. Disease presentation ranges from an asymptomatic finding on a routine full blood count to an acutely unwell patient suffering the clinical consequences of severe anaemia, neutropenia or thrombocytopenia. The cytopenia may be primary or secondary to underlying infectious, immune or malignant processes. Thrombotic thrombocytopenic purpura (TTP) is a distinct, rare but potentially life-threatening entity that classically but not invariably presents with a pentad of acute onset haemolytic anaemia, thrombocytopenia, neurological symptoms, renal impairment and fevers. Autoimmune cytopenias have formed a recognised diagnostic entity for over 150 years yet continue to present a challenge in medical practice due to heterogeneity in clinical presentation and triggering factors, an incomplete understanding of underlying pathophysiology and a lack of evidence-based therapeutic approaches.
Key points
Autoimmune disease may manifest as deficiency of any blood lineage, in isolation or in combination
Autoimmune cytopenias and thrombotic thrombocytopenic purpura (TTP) may be the primary manifestation of disease or reflect underlying malignancy, infection, exposure to certain medications or multisystem disease
The immune cytopenias range in severity from isolated laboratory finding to life-threatening rapidly progressive disease
Liaison with haematology and rapid initiation of plasma exchange is key in limiting morbidity/mortality in TTP
Autoimmune cytopenias are more common in pregnancy and women of child-bearing age and often represent a diagnostic and management challenge
Background
Autoimmune cytopenias are an important, treatable cause of deficiency of one or more of the blood lineages. They occur due to immune system dysregulation resulting in loss of immune tolerance to glycoproteins expressed on the surface of blood cell subsets. Autoantibody formation occurs, and both peripheral red cells, white cells or platelets and their bone marrow progenitors are antibody-bound and subsequently phagocytosed and cleared by macrophages.1,2 Additionally, cellular immunity is disturbed and shifted to a proinflammatory immune response.3 In thrombotic thrombocytopenic purpura (TTP), autoantibodies are targeted to and deplete ADAMTS13, the von Willebrand factor (VWF) cleaving protease. Rarer hereditary forms of TTP result from mutations in ADAMTS13, causing severe protease deficiency. Lack of ADAMTS13 results in failure to cleave ultra large multimers of VWF, leading to platelet aggregation and thrombotic microangiopathy. Autoimmune cytopenias are often associated with underlying disease, pregnancy and the postpartum period (Table 1).4
Secondary causes of immune cytopenias and TTP
Diagnosis of autoimmune cytopenia can be challenging. The differential for a low blood count is broad, and establishing an autoimmune aetiology requires careful incorporation of clinical history, examination and laboratory blood testing (Table 2). There is often no specific diagnostic test, making an autoimmune aetiology a diagnosis of exclusion. In this article we outline the clinical work-up, diagnosis and management of the common autoimmune cytopenias and TTP.
Clinical evaluation of the cytopenic patient
Autoimmune haemolytic anaemia
Autoimmune haemolytic anaemia (AIHA) affects both children and adults with an incidence of ∼1 per million.5 It is traditionally classified according to the temperature dependence of the causative autoantibody: warm antibodies cause ∼90% of AIHA in adults (∼75% in children) and are IgG antibodies that bind most avidly at body temperature, resulting in extravascular, primarily splenic, haemolysis. Cold autoantibodies are IgM antibodies that bind most avidly at around 4°C, leading to activation of complement and either intra- or extra-vascular haemolysis (cold haemaglutinin disease [CHAD]).6 Evans syndrome is a variant of AIHA and describes the occurrence of two or more immune cytopenias, most often AIHA and immune thrombocytopenic purpura (ITP).
In both types of AIHA, patients may present with classical symptoms of anaemia, such as breathlessness and fatigue. Extravascular haemolysis leads to signs of jaundice, dark urine +/- splenomegaly, and intravascular haemolysis results in haemoglobinuria. CHAD patients may experience localised pain, cyanosis and, in extreme cases, gangrene in the fingers, toes or ear lobes.7 In approximately 50% of cases AIHA is primary, with no clear underlying disease association. Secondary AIHA is most often seen in the context of B cell malignancy, autoimmune disease or drugs.8
Laboratory investigations in AIHA demonstrate a classic haemolytic picture, with a low, usually normocytic, haemoglobin, raised lactate dehydrogenase (LDH), raised bilirubin and reticulocytosis. The blood film demonstrates spherocytosis and polychromasia, with red cell agglutination seen in CHAD (see Fig 1a). The key diagnostic test is a positive direct antiglobulin test (DAT), in the absence of other obvious cause of haemolysis. The DAT detects antibody or complement bound to red cell membrane: the patient's red cells are washed and then specific antibodies to IgG and/or complement are added. If IgG and complement are bound to the red cell membrane, agglutination results and the test is positive. Further investigation of AIHA is aimed at excluding a secondary cause and should include viral serology, protein electrophoresis and an autoimmune screen.8 A computed tomography (CT) chest/abdomen/pelvis should be considered to rule out underlying lymphoid malignancy. Patients with CHAD aged ≥60 years where features of the history, examination, full blood count (FBC) or blood film suggest marrow infiltration should undergo a bone marrow biopsy.8
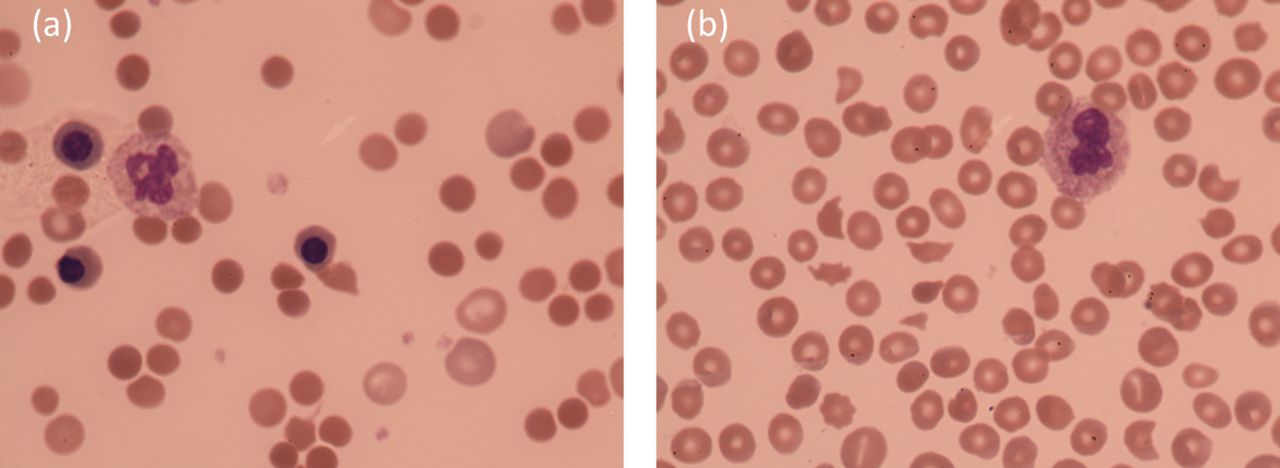
Panel of blood films; (a) AIHA, (b) TTP.
Treatment of AIHA aims to reduce symptoms and suppress autoantibody production. Where haemolysis is rapid or severe, and the patient is acutely symptomatic, transfusion may be required. Cross-matching blood may be difficult due to the presence of a pan-reacting antibody.9 A warm antibody renders exclusion of an underlying alloantibody impossible without further investigation and consequently leads to delay to transfusion. The patient should be discussed with the blood bank team, and if urgent blood is required, a patient can receive ‘least incompatible’ (ABO, Rh and Kell matched) blood. If cold agglutinins are present, interference can be overcome by keeping the blood warm during transfer from patient to laboratory, and a blood warmer is required during transfusion to help prevent acute intravascular haemolysis.9 The mainstay of therapy is glucocorticoids, with gastric protection, tapered slowly with monitoring of the haemoglobin and DAT.10 Second-line therapies include splenectomy, IvIg, immunosuppressants (azathioprine, cyclosporine), or rituximab (anti-CD20). Folic acid 5 mg once daily is given to support compensatory red cell production by the bone marrow to match the extent of haemolysis.
Autoimmune neutropenia
Autoimmune neutropenia (AIN) is a rare, heterogeneous group of diseases characterised by production of IgG autoantibodies directed against neutrophils, resulting in phagocytosis and splenic clearance.11 It may be primary or secondary (Table 1). Patients may be entirely asymptomatic or suffer recurrent infection, although the risk of infection is lower than observed with chemotherapy-induced neutropenia. Where infections occur, these are usually limited to the mucosa or skin and characterised by an absence of pus formation.11
Laboratory testing is valuable in confirming the severity of neutropenia and for monitoring the disease. Once neutrophils are <0.5 × 109/L there is a significantly increased risk of infection. However, assays for detection of anti-neutrophil antibodies are generally not useful due to poor sensitivity. As such, AIN is commonly a diagnosis of exclusion.12
Management depends on patient age and frequency of infection. Primary AIN is most common in infants, and is typically benign and self-limiting, requiring observation only. Adults with AIN may require antibiotic prophylaxis and granulocyte colony-stimulating factor during infections. If AIN is severe, or associated with other autoimmune disease, treatment with steroid, IvIg, or immunosuppressants such as rituximab is indicated. Here, ensuring a correct diagnosis and close observation are paramount, as the drugs may initially worsen the neutropenia.
Autoimmune thrombocytopenia
Autoimmune thrombocytopenia (ITP) is an autoimmune disorder characterised by an isolated low platelet count and mucocutaneous bleeding. The incidence is 1–3 per 100,000 per year, of which ∼50% of cases occur in children.13 ITP may present in any gender and at any age, peaking at age 5 and in the elderly. Adult-onset and childhood-onset ITP present differently.4 Children present with sudden onset of purpura or petechiae, often shortly following a viral illness, and 80% resolve within 1 year.14 In adults, the onset is insidious, and the disease often takes a chronic course.15 Bleeding risk is related to platelet count, with those with platelet counts of <10 × 109/L at greatest risk. An underlying cause for ITP is identified in 20% (Table 1).1
Laboratory investigations demonstrate an isolated low platelet count <100 × 109/L. Further investigations are indicated to exclude a secondary diagnosis. Bone marrow biopsy may be performed to exclude myelodysplastic syndrome and shows abundant megakaryocytes and otherwise normal cellular morphology.16
The aim of treatment in ITP is to achieve a platelet count which maintains haemostasis, rather than one in the normal range. Most patients do not require treatment if the platelet count is >30–50 × 109/L. Important supportive measures include stopping drugs that impair platelet function, optimising blood pressure control and controlling menstrual bleeding. Patients are educated to recognise signs of a low platelet count, to avoid contact sports and how to get emergency help.
Treatment for ITP is divided into emergency acute treatment and management of chronic ITP.17 In the event of a catastrophic bleed such as an intracranial haemorrhage, platelet transfusion should be administered, along with intravenous methylprednisolone, IvIg and tranexamic acid. Treatment of chronic ITP is tailored to the individual and based on age, comorbidity, bleeding history, and risk of treatment-related side effects. Criteria for treatment include active bruising or bleeding, high risk of bleeding, rapidly falling counts or the need for an invasive procedure. High-dose corticosteroid with a rapid taper is the most common first-line therapy. However, while ∼66% of patients respond, this is durable in only 10–30%. Second-line therapies are heterogeneous, and include splenectomy, rituximab, long-term immunosuppression with mycophenolate mofetil / azathioprine, or the thrombopoietin receptor agonists eltrombopag and romiplostim. Splenectomy is associated with the highest cure rate of 60% at 5 years, but risks peri and postoperative complications. Indium-labelled platelet sequestration studies scan can help identify those most likely to benefit.18
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura (TTP) is a rare disorder, with an incidence of 6 per million per year in the UK.19 The diagnosis should be considered if there is thrombocytopenia and microangiopathic haemolytic anaemia (MAHA) with schistocyes (Fig 1b), before the classic pentad including neurological signs, renal impairment and fever arises. Untreated, mortality approaches 90% and as such, awareness, early recognition and immediate management are key to ensuring patient survival. As with the autoimmune cytopenias, TTP can be primary, or secondary to underlying disease.
Unlike AIHA, DAT is negative and clotting tests should be normal. Troponin is elevated in 50% and is a poor prognostic marker. The key confirmatory diagnostic test is the ADAMTS13 protease level with severe disease classed as <5% activity. The presence of antigen, neutralising and non-neutralising anti-ADAMTS13 autoantibodies helps identify an autoimmune aetiology.20 Serial measurements can be helpful in evaluating disease trajectory.
Immediate plasma exchange (PLEX) (within 4–8 hours) is key to management. This has helped reduce mortality from >90% to 10–20%.20 PLEX reduces the auto-antibody, the pathological ultra-large von Willebrand multimers and replaces ADAMTS13. Immunosuppression with high-dose steroid should be started concomitantly alongside other supportive care measures including red cell transfusion to correct anaemia, folic acid to support marrow function, protein pump inhibitors for gastric protection and thromboprophylaxis with low-molecular-weight heparin and aspirin once platelet count >50 × 109/L. Platelet transfusions are contraindicated unless life-threatening haemorrhage occurs. In refractory cases or where there is neurological or cardiac involvement, B cell depletion with rituximab is indicated.20
Conclusions
The autoimmune cytopenias and TTP are an important differential in any patient presenting with a cytopenia, as effective treatments are available. Incorporation of clinical history, examination and laboratory investigations is key in establishing the diagnosis and ruling out underlying secondary causes. Research is ongoing to investigate the underlying mechanisms of immune dysregulation and there is hope that this will continue to translate into advances in diagnosis and management.
- © Royal College of Physicians 2018. All rights reserved.








{kind=link}