
Introduction
Sudden cardiac death (SCD) in the young and the athletes is a devastating event. It occurs unexpectedly in apparently healthy people, often during maximal, vigorous cardiac performance. In 1980 we observed a series of cases and perceived that an investigation covering all the victims in the Veneto Region of Italy might help to clarify the puzzle. Since then, in the time interval 1980–2013 we studied 650 consecutive sudden death (SD) cases in the young and athletes.1,2 Collaboration with pathologists and forensic doctors was crucial. The aim was clear: to assess the epidemiology of the phenomenon and to identify the ‘killer’ diseases – in particular, to identify morbid conditions which could be genetically transmissible, ie hereditary. Is this catastrophe predictable because written in our DNA? Is it possible to identify subjects at risk? The method of investigation was autopsy with gross, histologic and, latterly, molecular investigation, together with clinical correlations. Since in Italy eligibility for sport activity requires ECG, the review of ECG records played a fundamental role by detecting even subtle abnormalities which could be of help to improve sensitivity and specificity of in vivo diagnosis and disqualify the subjects from strenuous sport activity. As far as definition of SD, we adopted the old definition of Morgagni ‘…and here with the name of sudden death we mean the death that, expected or unexpected, kills the man abruptly.’3
Mechanisms of sudden death
The primary mechanism of cardiopulmonary arrest may be cerebral, respiratory or cardiovascular.4 Subarachnoidal haemorrhage, due to rupture of a congenital berry aneurysm of the Willis circle, is the main cerebral mechanism, with respiratory pacemaker stoppage in the brain stem. Allergic bronchial asthma, with bronchospasm and mucoid plugs obliterating the air pathways and preventing the subject from breathing, is the principal peripheral respiratory mechanism.
In our initial series of three hundred cases, 5% were cerebral and 4% respiratory death, whereas the large majority (91%) were cardiovascular. The latter, according to the pathophysiologic mechanism, were mechanical (7%) with the cardiac arrest as the consequence of circulation blockage due to mechanical obstacle (cardiac tamponade by haemapericardium, pulmonary thromboembolism), or electrical because of arrhythmias (93%).4 The arrhythmic mechanism is generally due to ventricular fibrillation, which is the greatest challenge of modern cardiovascular medicine. John MacWilliam (1857–1937) first realised that:Sudden cardiac failure does not usually take the form of a simple ventricular standstill in diastole… It assumes, on the contrary, the form of violent, though irregular and incoordinated, manifestation of ventricular energy. Instead of quiescence, there is a tumultuous activity, irregular in its character and wholly ineffective as regards its results.5 Moreover this:…is attended with disastrous results: an immediate abolition of the normal beat, and the occurrence of a wildly incoordinated, arrhythmic contraction of the ventricular muscle (fibrillary contraction or heart delirium), attended by a great and rapid fall of blood-pressure, and, in the higher mammals…, speedy death.5
Cardiac etiologies of sudden death in the young
In our investigation we found that the substrate accounting for SCD is located in pathology of the structural components of the heart, namely in the aorta and pulmonary artery, coronary arteries, myocardium, valves, conduction system and ion channels.
Aorta
Aortopathy in the setting of bicuspid aortic valve, with aortic dissection and rupture, accounted in our experience for 3% of SD cases (Fig 1). The aortic wall presented at histology medionecrosis and elastic fragmentation, like those observed in Marfan syndrome.6 In contrast to the latter, bicuspid aortic valve aortopathy is rarely hereditary. The etiology and pathogenesis of the aortic wall degeneration is still a mystery.
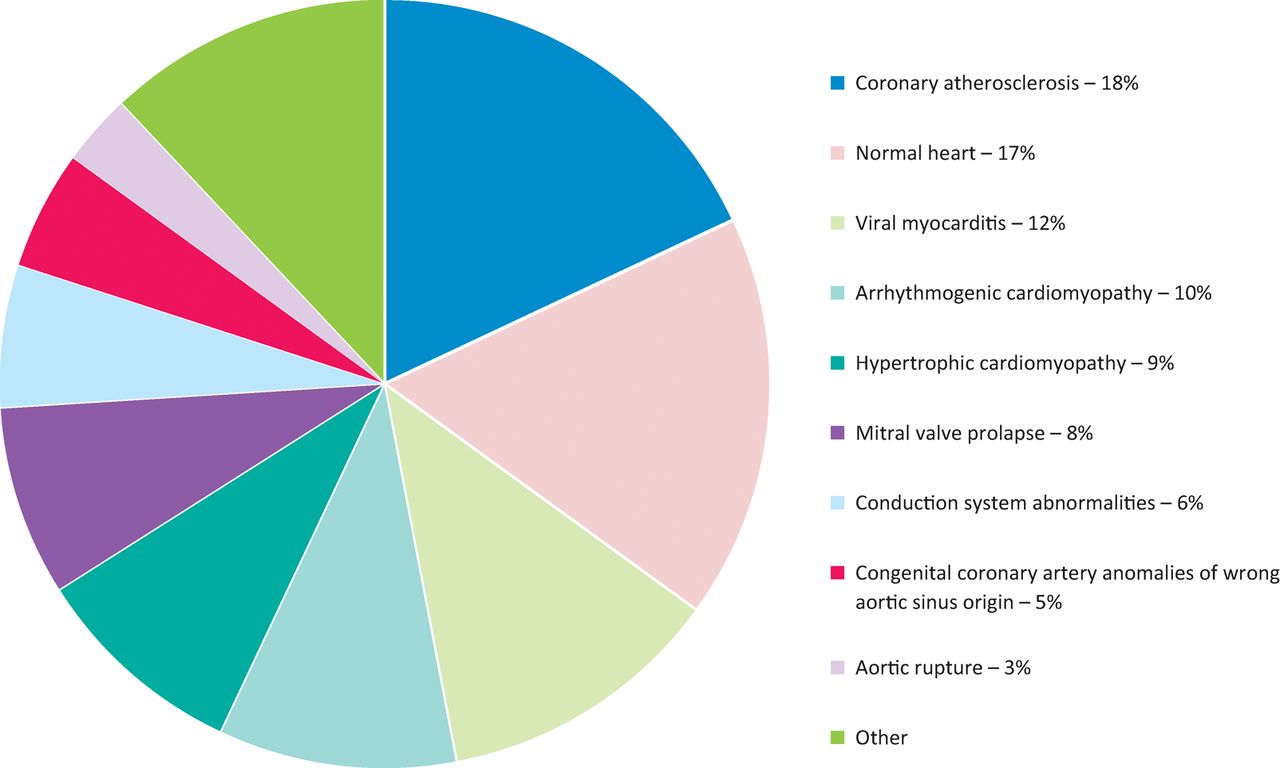
Etiologies for sudden cardiac death in a series of 650 consecutive sudden death cases in the young and athletes in the Vento region 1980–2013.
Coronary arteries
Coronary atherosclerosis is the leading cause of SCD even in the young and accounted for 18% of deaths (Fig 1). Coronary thrombotic occlusion in the young, in contrast to adults and the elderly where it is observed in nearly 70–80% of coronary SCD cases, accounted only for one-third of cases and was mostly due to endothelial erosion. The remaining cases presented a single obstructive atherosclerotic plaque, located in the proximal descending coronary artery in the absence of occlusive thrombosis.7 We were able to prove in young subjects dying suddenly during Holter monitoring that the final mechanism is transient coronary occlusion by vasospasm, with ventricular fibrillation triggered by myocardial reperfusion.8 Thus, coronary SD by atherosclerosis in the young may be either structural (coronary thrombosis) or functional (coronary vasospasm).
Regarding hidden congenital coronary anomalies, the origin of a coronary artery from a wrong (opposite) sinus, with initial course in between the aorta and pulmonary artery and slit-like lumen, was observed in 5% of all SDs in the young 9,10 (Fig 1). Cardiac arrest occurs during prolonged effort, clearly because of a discrepancy between coronary blood flow request and supply.
Myocardium
Myocarditis is a leading cause (12%) of SCD in the young (Fig 1). With the employment of molecular techniques, it is possible not only to establish that the myocardium is inflamed, but also to detect the causative microorganisms. Coxsackie virus (an RNA enterovirus) was found to be the most frequent malignant cardiotropic agent.2,4,11
Hypertrophic cardiomyopathy, with a prevalence of 1:200–1:500 in the general population, accounted for 9% of SCD victims (Fig 1). The heart presents with asymmetrical hypertrophy of the left ventricle, myocardial disarray at histology and ischaemic scars due to impaired coronary microcirculation reserve.12 Myocardial bridge is part of the phenotypic spectrum of hypertrophic cardiomyopathy and can account in selected cases for ischemia.13 It is a hereditary disease due to mutations of genes encoding sarcomeric proteins (‘sarcomeric disease’).
Arrhythmogenic cardiomyopathy is a malignant disease affecting mainly the right ventricle with fibrofatty replacement of the myocardium.14,15 The prevalence of the disease ranges from 1:2000 to 1:5000 in the general population. It accounts for 10% of SCD in the young (Fig 1). It is also hereditary and it was proven to be the consequence of mutations of genes encoding cell junction proteins (‘desmosomal disease’).16,17 Nowadays, non-ischemic scars within the myocardium may be detected by cardiac magnetic resonance with gadolinium late enhancement.18 Electroanatomic mapping helps to reveal electrical scars (‘electric silence’), either in the right or left ventricle,19 which are the origin of life-threatening re-entry ventricular arrhythmias.
Valve disease
Bicuspid aortic valve is associated with degenerative aortopathy and is at risk of aortic dissection and rupture with cardiac tamponade and mechanical cardiac death (see above). Bicuspid aortic valve is found in 1–2% of the general population.
Mitral valve prolapse has an estimated prevalence of 2–3% in the general population. Mitral valve prolapse explains SCD in 8% of cases (Fig 1). It is an arrhythmic phenomenon and occurs especially in young women with palpitations and recorded ventricular arrhythmias.20 It has been demonstrated that this electrical instability originates from myocardial fibrosis in the mitral papillary muscles and mural myocardium, most probably due to mechanical stress because abnormal annular attachment of the leaflet. The fibrosis is easily detectable by cardiac magnetic resonance with late enhancement and may be the site of ablation.21
Conduction system
Disease of the conduction system, with abnormal propagation of the electrical impulse, accounts for 6% of juvenile SCD (Fig 1). The most frequent culprit disease is Wolff–Parkinson–White syndrome where an accessory bundle (‘Kent fascicle’) connects the atria to the ventricular myocardium, outside the conduction system, leading to ventricular preexcitation. This thin pathway (<200 microns thick) consists of working myocardium with fast conduction and a low refractory period. Paroxysmal atrial fibrillation may transform into ventricular fibrillation and SCD, since the anomalous fascicle may conduct 1 to 1 to the ventricles.22 It has been estimated a prevalence of 1:1000 individuals with ECG pre-excitation patterns in the general population.
Sudden cardiac death and normal heart
The heart may stop instantaneously because of ventricular fibrillation even in the absence of a morphological substrate (so called ‘mors sine materia’); this accounts for 17% of young SCD victims in our experience (Fig 1).1,2 Rudolf Virchow in 1894 said ‘any anatomic modification is material, but is any material modification anatomic? Why not molecular…in the setting of a normal structure?’ The electrical instability resides in abnormal depolarisation–repolarisation of the myocyte, due to alterations of ionic pumps, currents and receptors of Ka+, Na+ and Ca++. Most of these abnormalities are evident at the ECG tracing.
Long QT syndrome is a genetic disorder due to mutations in genes encoding the proteins that govern Ka+ and Na+ currents and account for prolonged depolarisation. An estimated LQTS prevalence of 1:2000 in newborns has been reported.
A short QT is featured by acceleration of repolarisation, mostly due to mutations of proteins controlling K+ current. The exact prevalence of short QT is unknown.
Brugada syndrome is characterised by non-ischemic ST-segment elevation, due to mutations of SCN5A gene, coding a sodium channel protein at the myocyte sarcolemma. Prevalence of Brugada syndrome in the general population ranges from 1:1000 in Asia to less than 1:10,000 individuals in Europe and America.
Catecholaminergic polymorphic ventricular tachycardia is due to abnormal release or uptake of the Ca++ from the smooth sarcoplasmic reticulum. The prevalence of the disease is estimated at 1:10000. Mutations of the ryanodine receptor II or calsequestrin (CASQ2) genes account for dominant and recessive forms, respectively. Oddly enough, in these morbid entities the basal ECG is normal and during emotion or effort it becomes abnormal with polymorphic ventricular arrhythmias.Table 1 summarises the cause of SCDs from various pathological reports.1,23–27
Causes of sudden cardiac death in the young (autopsy-proven)
A not negligible proportion of SCD cases (up to 50% of cases) present with a structurally normal heart at autopsy.5 The great variability in the prevalence of ‘normal’ heart in autopsy studies in the literature can reflect the use of non-uniform protocols of investigation at autopsy, as well as the overestimation of the so-called borderline findings of uncertain significance. In the setting of proven autopsy-negative SCD, comprehensive or targeted (RyR2, KCNQ1, KCNH2, and SCN5A) ion channel genetic testing may be considered in attempt to establish probable cause and manner of death and to facilitate the identification of potentially at-risk relatives.28–30 In particular, genetic testing is recommended if circumstantial evidence points toward a clinical diagnosis of LQTS syndrome or CPVT specifically (such as emotional stress or drowning as the trigger of death).
The key question is whether molecular autopsy should be applied to all SCD victims with normal hearts or if it should be used only when clinical evaluation of family members has generated some concern that a genetic cardiac disease might be present.28 In fact, the advantage of pursuing genetic screening of the unexplained SCD victim over performing clinical evaluation of family members has not been conclusively demonstrated.31 Last, but not least, the impact of ‘uncertain results’ of genetic testing, represented by the identification of variants that cannot be conclusively defined as ‘pathogenic,’ has not been evaluated. In the real world, it has been calculated that 10–15% of samples from unexplained SCD victims carry a mutation in KCNQ1, KCNH2, or SCN5A. The addition of the screening of the RyR2 gene, for mutations that cause CPVT, may increase the yield of molecular autopsy by an additional 5–10%, thus bringing up to 15–25%.32,33
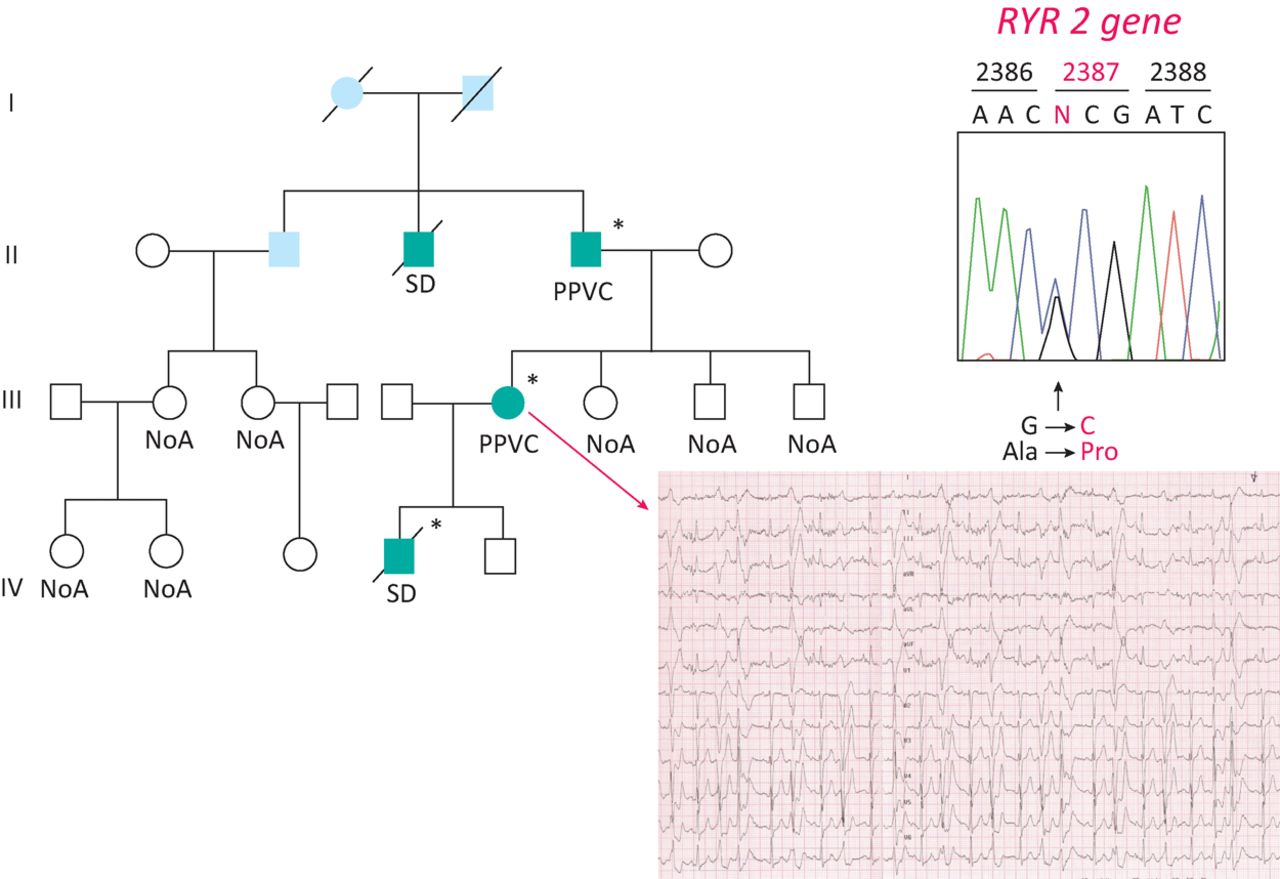
At the present time, it seems reasonable to advocate for a combined approach, with parallel clinical assessment of the family members of unexplained SCD victims and molecular autopsy on the proband. In case of a ‘positive’ genetic test in the unexplained SCD victims, cascade genetic screening may be extended for the same ‘variant’ (Fig 2). Screening of first degree relatives of SCD victims identifies a hereditary arrhythmogenic syndrome in around 50% of families, thereby providing a likely cause of death and identifying surviving relatives at risk from the same fate.34
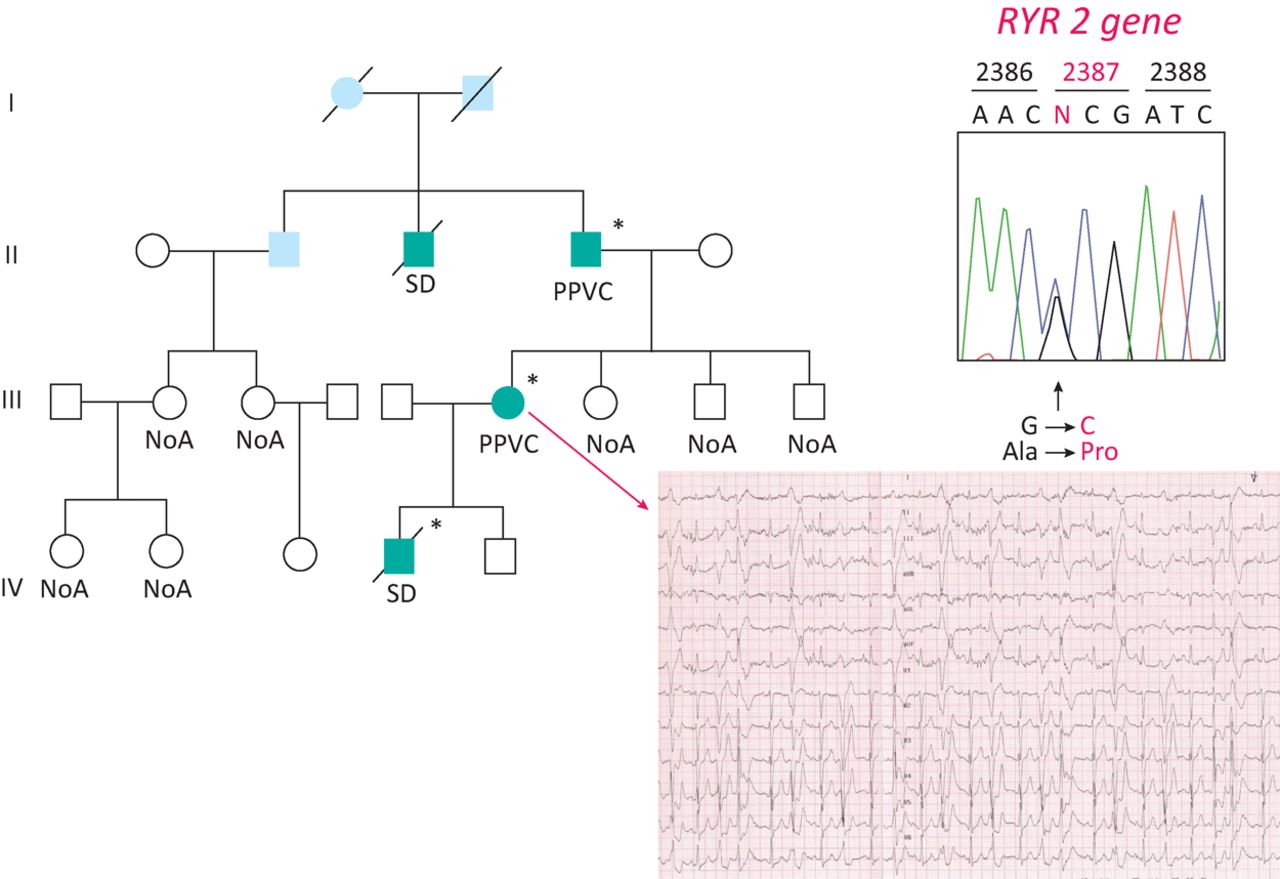
Molecular autopsy in a boy with SCD and normal heart. A missense mutation was found in the gene encoding the ryanodine receptor II, governing the release of Ca++ from smooth sarcoplasmic reticulum for excitation-contraction coupling. The mother, during stress test, developed ventricular arrhythmias and was found to be carrier of the same gene mutation.
As new technologies are reducing the cost of genotyping, it is expected that the prices may rapidly decrease, thus making the postmortem analysis more widely used. With the genome-wide investigation technique, it is foreseeable that eventually a genetic explanation will be found in many other cases of ‘mors sine materia.’ However, after the initial emphasis, caution is needed due to the difficult interpretation of the pathogenic significance of gene mutations in the absence of family history and cascade genetic screening.35,36
Sudden death in athletes
Effort and sporting activity may precipitate ventricular fibrillation and sudden death by unmasking hidden cardiac disease. Our epidemiological study in the Veneto Region established that overall the cumulative incidence of SCD in the young (<35 years of age) is 1/100,000/year (2.3/100,000/year in the athletes vs 0.9/100.000/year in non-athletes). With respect to arrhythmogenic cardiomyopathy, the risk of sudden death is 5.4 higher in athletes than in non-athletes.37
In Italy, eligibility for competitive sport is mandatory by law, and includes visits with history taking, physical examination and basal ECG. In case of doubt, stress test ECG and echo is compulsory. Box 1 summarises the various levels of cardiological screening: first level investigation includes history, physical examination and 12-lead ECG, second level consists of non-invasive tools, and the third level, when deemed necessary, involves the use of invasive tools, including endomyocardial biopsy. Genetic screening, both in the proband and first degree relatives, is strongly indicated when the subject is found to be affected by the phenotypic expression of a genetic disorder or presents a normal heart. Overall, 50% of SCD in the young is ascribable to genetic disorders.
How to detect diseases at risk through cardiological screening
The benefits of the Italian system have been demostrated by the sharp decline in sudden death in athletes in Italy after the introduction of obligatory ECG for eligibility, with the incidence dropping by 90%.38 This was particularly true for hypertrophic cardiomyopathy. In contrast with the USA, where the ECG is not compulsory for eligibility and the rate of hypertrophic cardiomyopathy as cause of sudden death is 26%, the rate in Italy is only 2%, because with our protocol athletes with hypertrophic cardiomyopathy are easily detected, disqualified and protected.39
However, we are well aware that disqualification is not a panacea to prevent sudden cardiac death. Basal ECG is quite effective and even specific to diagnose hypertrophic cardiomyopathy, arrhythmogenic cardiomyopathy and ion channel diseases, but lacks sensitivity and specificity in coronary artery disease, both acquired and congenital, and valve diseases. Therefore, there are cardiovascular causes of SCD that still escape the preparticipation screening, due to a normal 12-lead ECG in asymptomatic athletes. In these circumstances, due to the absence of previous symptoms or signs, there was – correctly – no reason to proceed to a second- or even third-level investigation to confirm the eligibility to sport activities. Moreover, we know that the role of screening in reducing sudden death rates in athletes is a very contentious area and that the cost-efficacy of this approach is certainly a matter of debate especially in the general population.40 For these reasons, in many cases, only prompt and adequate resuscitation manoeuvres, including the use of public access defibrillators, may save athletes’ lives. Thus, promotion of secondary prevention by dissemination of automated external defibrillator and of the culture of resuscitation to professionals or even lay people trained to use the device can make the difference in such cases.41 This appears to be the case even in countries where cost-effectiveness considerations for preparticipation screening, including the ECG, do not limit access to further cardiologic testing.42 High survival rate (61–87%) has been reported across different studies if the SCD victim was found with a shockable rhythm and received early defibrillation.43 Noteworthily, since the majority of SCA events occur in the home, it has been demonstrated that people who purchase an automated external defibrillator for their home, even without previous experience, are able to use the device successfully in both adults and children with high survival rate.44
Modern standards based on new and emerging research have been proposed to distinguish physiologic cardiac adaptations in athletes from findings suggestive of underlying pathology, namely the European Society of Cardiology criteria45 and the ‘Seattle criteria’,46 which have recently been updated.47
A variety of ECG alterations have been reported in patients with hypertrophic cardiomyopathy (eg T-wave inversion in inferior-lateral leads, pathological Q waves, left axis deviation).
The most common ECG abnormalities in arrhythmogenic cardiomyopathy include prolonged QRS ≥110 ms, inverted T-waves in the right precordial leads (V1–V3), and late potentials by signal averaged. Epsilon waves and ventricular arrhythmias such as premature ventricular contractions or non-sustained / sustained ventricular tachycardia are common, typically with a left bundle branch block pattern.
An ECG showing a characteristic delta wave provides the diagnosis of ventricular pre-excitation.
A QT interval corrected for heart rate (QTc) ≥480 ms is required to establish the diagnosis of long QT syndrome. Current ESC guidelines propose a QTc ≤340 ms to diagnose short QT syndrome.
Catecholaminergic polymorphic ventricular tachycardia shows normal resting ECGs. Exercise stress testing or emotions elicit the appearance of the pathognomonic polymorphic ventricular tachycardia.
The Brugada syndrome is diagnosed when ST-elevation is observed in at least one right precordial lead (V1 and V2).
Dissemination of automatic external defibrillators is extremely useful, effective and strongly recommended. They should be scattered and available for prompt use in sport grounds when necessary. Their availability is now compulsory by law in Italy even for non-competitive sport.
Therapy
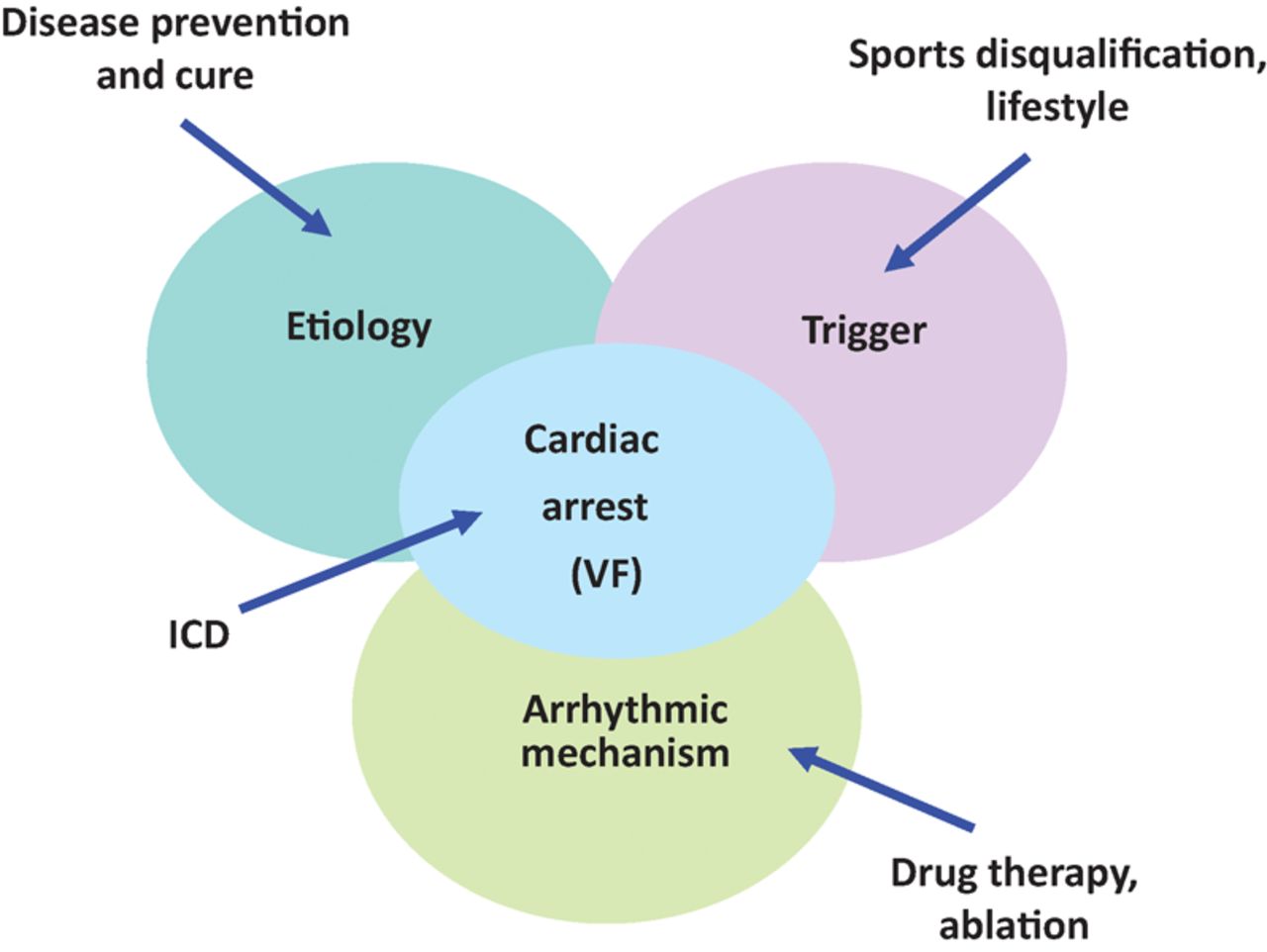
Prevention of sudden death in the young is feasible (Fig 3).48 Lifestyle and sport disqualification represent elementary, effective approaches to prevent the trigger. Antiarrhythmic drugs and ablations of re-entry circuits are widely employed with controversial efficacy. Implantable cardioverter defibrillators are life-saving and allow resuscitation from cardiac arrest by ventricular defibrillation. The entirely subcutaneous defibrillator (S-ICD) represents a valid alternative to the transvenous device and can provide substantial advantages, especially among some subgroups of patients (ie after device infection, in young patients with an active lifestyle and a long life expectancy, and in arrhythmogenic syndromes). However, it is fundamental to choose patients that can benefit the most. Due to its characteristics S-ICD appears suited to young patients with inherited genetic arrhythmogenic syndromes (Brugada, long and short QT, or early repolarisation) where clinical arrhythmias are polymorphic VT or VF and the risk of bradycardia and monomorphic VT is very low.49
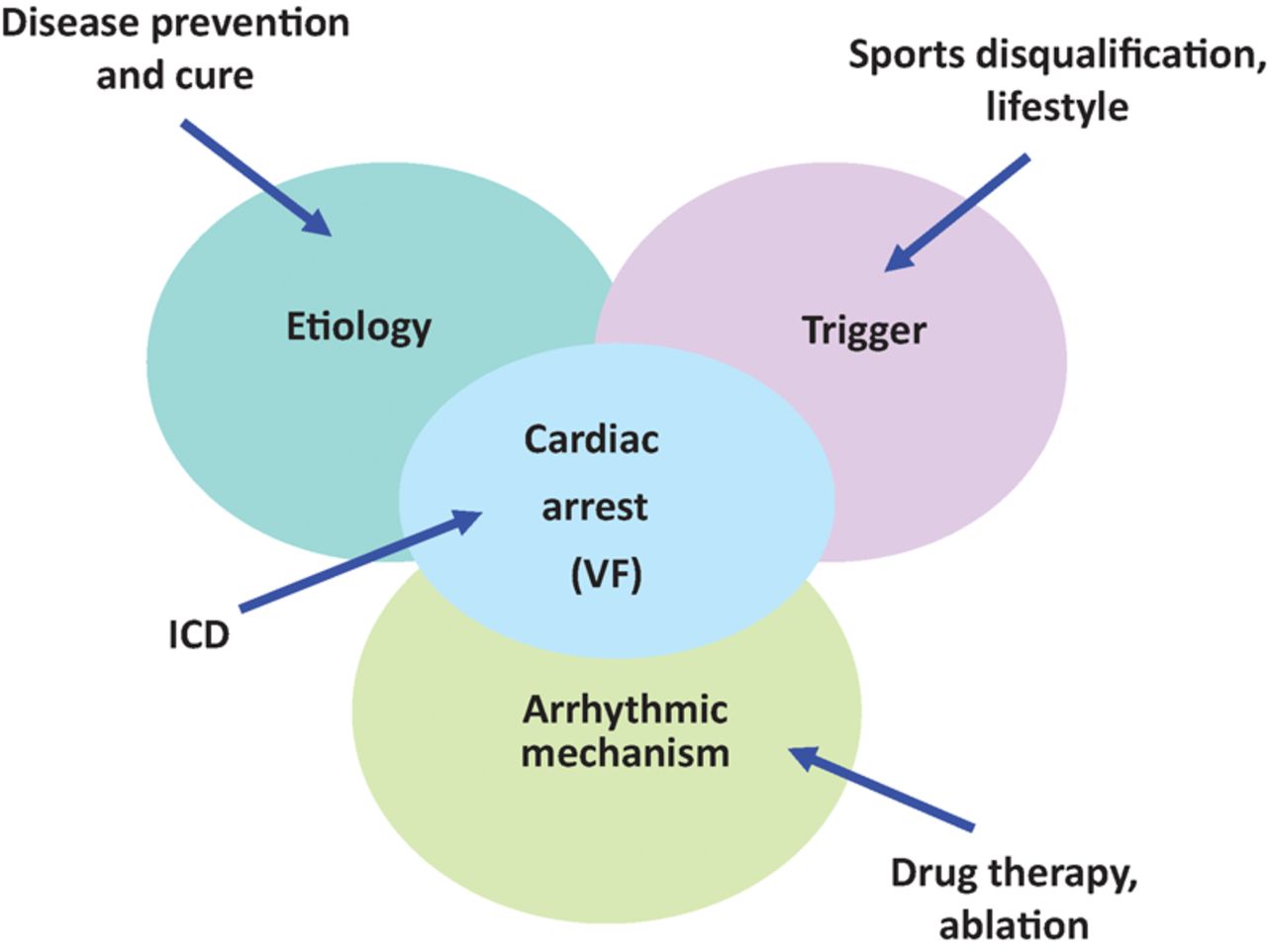
Multiple approaches to prevent SCD in the young, addressing the trigger, the arrhythmic mechanism, the arrest by ventricular fibrillation through defibrillator (ICD) and the etiology, the latter to find a cure of the disease.
However, these types of therapy are clearly palliative and symptomatic. Ideally the objective is to find a cure for the disease. As far as arrhythmogenic cardiomyopathy, transgenic mice have demonstrated that the disease is genetically determined and that the phenotypic expression is not present at birth.50,51 The main goal should be to stop the onset of the disease in childhood and the progression with time. Studies in zebrafish have suggested new targets for drugs able to block the progression of the disease.52
Human-induced pluripotent stem cells from dermal fibroblasts is quite a promising method to obtain mature cardiomyocytes of the patient themselves and to recapitulate the arrhythmogenic cardiomyopathy genotype pathophysiology for a personalised therapy.53
Gene therapy with the delivery of an Adeno-associated viral construct was found to be efficient in knock-in mice of CASQ2, a catecholaminergic polymorphic ventricular tachycardia gene, either at birth or in adults.54 Last but not least, genome editing techniques are on the therapeutic horizon, with the potential for the targeted correction of germline mutations. Recently, Ma et al55 described the correction of the heterozygous MYBPC3 mutation in human preimplantation embryos with precise CRISPR–Cas9-based targeting accuracy and high homology-directed repair efficiency by activating an endogenous, germline-specific DNA repair response. The authors were able to achieve a high yield of homozygous embryos carrying the wild-type MYBPC3 gene without evidence of off-target mutations. This approach has potential to be used for the correction of heritable mutations in human embryos by complementing preimplantation genetic diagnosis, although the reproducibility of the technique with other heterozygous mutations remains to be demonstrated before clinical applications.
Conclusion
SCD in the young is due to a wide spectrum of hidden, poorly symptomatic diseases. There are a number of options for early in vivo identification of subjects at risk, including use of ECG and clinical imaging. SCD prevention has to be approached by an interdisciplinary team (cardiologists, geneticists, pathologists), the clinicopathologic correlation method being the polar star (‘translational medicine’). Considering that 50% of SCD cases are genetic in origin, it is no longer justifiable to fail to genotype victims and to screen first degree relatives if mutations are detected. This approach should become part of the routine postmortem study of SCD cases.56
The study of SCD moved from the traditional postmortem dissection to molecular autopsy. Nonetheless the game still starts from the anatomical theatre, ‘the place where death enjoys to save lives’.
The effective treatment has to focus the molecular mechanisms that are involved in the disease etiopathogenesis. Cure of these hereditary cardiac diseases at risk of SCD is on the horizon. We all look forward to the time when arrhythmias will no longer be the only target to prevent SCD.
Consent
Consent to publish the details in Fig 2 was obtained at the time the family underwent genetic screening.
- © Royal College of Physicians 2018. All rights reserved.








{kind=link}
{kind=link}
{kind=link}