
ABSTRACT
The haematological malignancies are a heterogeneous group of neoplastic disorders, which lead to almost 10,000 deaths annually in the UK. Over the past 2 decades, there has been significant progress in our understanding of the pathological mechanisms underlying these cancers, accompanied by improvements in outcomes for some patients. In particular, advances in next-generation sequencing now make it possible to define the genetic lesions present in each patient, which has led to improved disease classification, risk stratification and identification of new therapeutic targets. Here we discuss recent advances in the genomic classification and targeted treatment of haematological malignancies, focusing on acute myeloid leukaemia. Multiple novel drug classes are now on the horizon, including agents that target overactive signalling pathways, differentiation therapies and immunotherapies. By combining molecular diagnostics with targeted therapy, the management of these diseases is set to change radically over the coming years.
Introduction
The haematological malignancies comprise a heterogeneous group of clonal neoplastic disorders arising from the haematopoietic and lymphoid tissues. They represent a broad spectrum of disease, from slowly progressive chronic leukaemias to acute leukaemias and high-grade lymphomas, which are among the most rapidly progressive of all human cancers. Approximately 25,000 cases of haematological malignancy are diagnosed annually in the UK, leading to almost 10,000 deaths.1 With the exception of childhood acute lymphoblastic leukaemia (ALL) and Hodgkin lymphoma, these are predominantly diseases of the elderly – in whom the incidence is rising.
In the last 2 decades, there has been major progress in understanding the genetic basis of these diseases, accompanied by substantial improvements in treatment for some subgroups of patients. The identification of the Philadelphia chromosome in chronic myeloid leukaemia (CML) and subsequent success of imatinib and other tyrosine kinase inhibitors targeting the BCR-ABL fusion kinase generated great optimism that molecularly targeted therapy would soon lead to cures for many other conditions. However, equivalent breakthroughs have not been forthcoming. It has become clear that, unlike CML, most cancers are not defined by a single molecular event but are instead genetically complex, making it highly unlikely that single-agent therapies will be curative unless they target generic cancer processes.
In this review, we discuss some recent advances in classification and treatment, focusing on acute myeloid leukaemia (AML) as an exemplar. Cytotoxic chemotherapy remains the standard of care for AML and has not changed substantially for over 40 years. As most patients are older and unfit for this therapeutic approach, mortality remains high. There has been no improvement in outcome for patients over the age of 75 years for at least 3 decades; in this group, median survival is only a few months.2 This is now beginning to change with a growing number of new treatments entering clinical trials.
From morphological towards genomic classification of disease
As with other cancers, the diagnosis and classification of haematological malignancies has traditionally been based on cellular morphology. For example, the 1976 French-American-British (FAB) classification divides AML into eight subtypes (M0-M7) based on the cell of origin and degree of maturation. Notably, acute promyelocytic leukaemia (APL, M3), which has an aggressive clinical course associated with life-threatening coagulopathy, has a distinct biology characterised by a chromosomal translocation involving the retinoic acid receptor-alpha (RARα) gene, and is uniquely responsive to treatment with all-trans retinoic acid (ATRA), as discussed later. However, with this exception, the link between morphology and clinical outcome is indirect. In contrast, classification based on molecular pathogenesis is likely to be more informative, provide better prognostic stratification and guide targeted treatment. Advances in genomic technologies now make this possible (see Table 1).
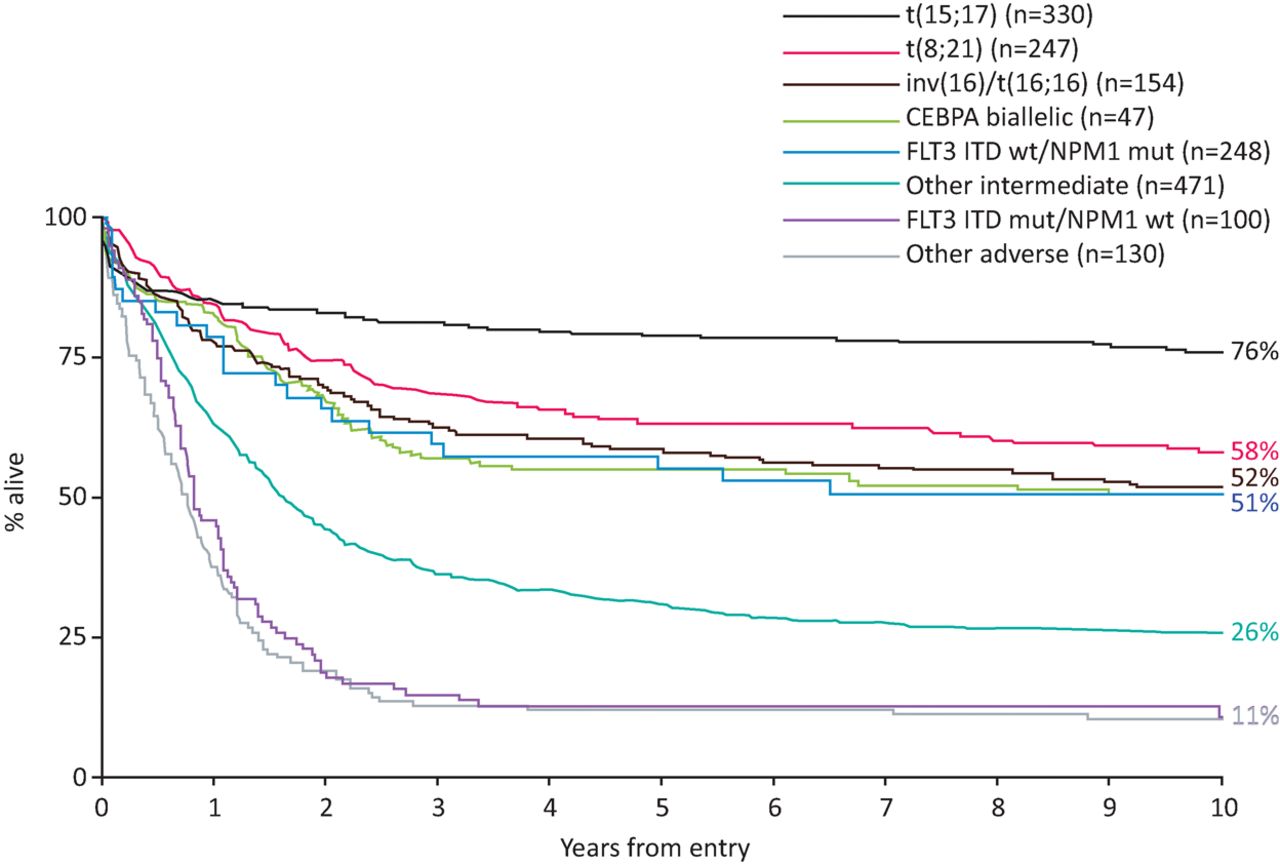
Cytogenetic abnormalities are relatively common in haematological malignancies and have been used routinely in prognostic stratification for 3 decades. Specific cytogenetic abnormalities in AML are associated with favourable- or adverse-risk disease (Fig 1).3–5 This informs decisions regarding treatment following induction chemotherapy. For example, in young patients with adverse-risk cytogenetics, trial evidence supports proceeding to allogeneic haematopoietic stem cell transplant (allo-HSCT) in the first remission.6 However, 40% of patients have a normal karyotype and fall into an intermediate-risk group, which has a diverse range of outcomes. There has been a considerable effort to identify molecular markers to further risk stratify this group of patients. An early study showed that in young patients with cytogenetically normal AML, mutations in NPM1 without FLT3 internal tandem duplication (FLT3-ITD) mutations, and in CEBPA were associated with a comparatively good outcome.7 In contrast, FLT3-ITD conferred a poor prognosis (Fig 1). Importantly, the benefit of allo-HSCT was limited to patients with FLT3-ITD and wild-type NPM1.4,7 In NPM1 mutant-AML, patients with persistent measurable residual disease (MRD) fare poorly and are also usually considered for an allo-HSCT in first complete remission.8
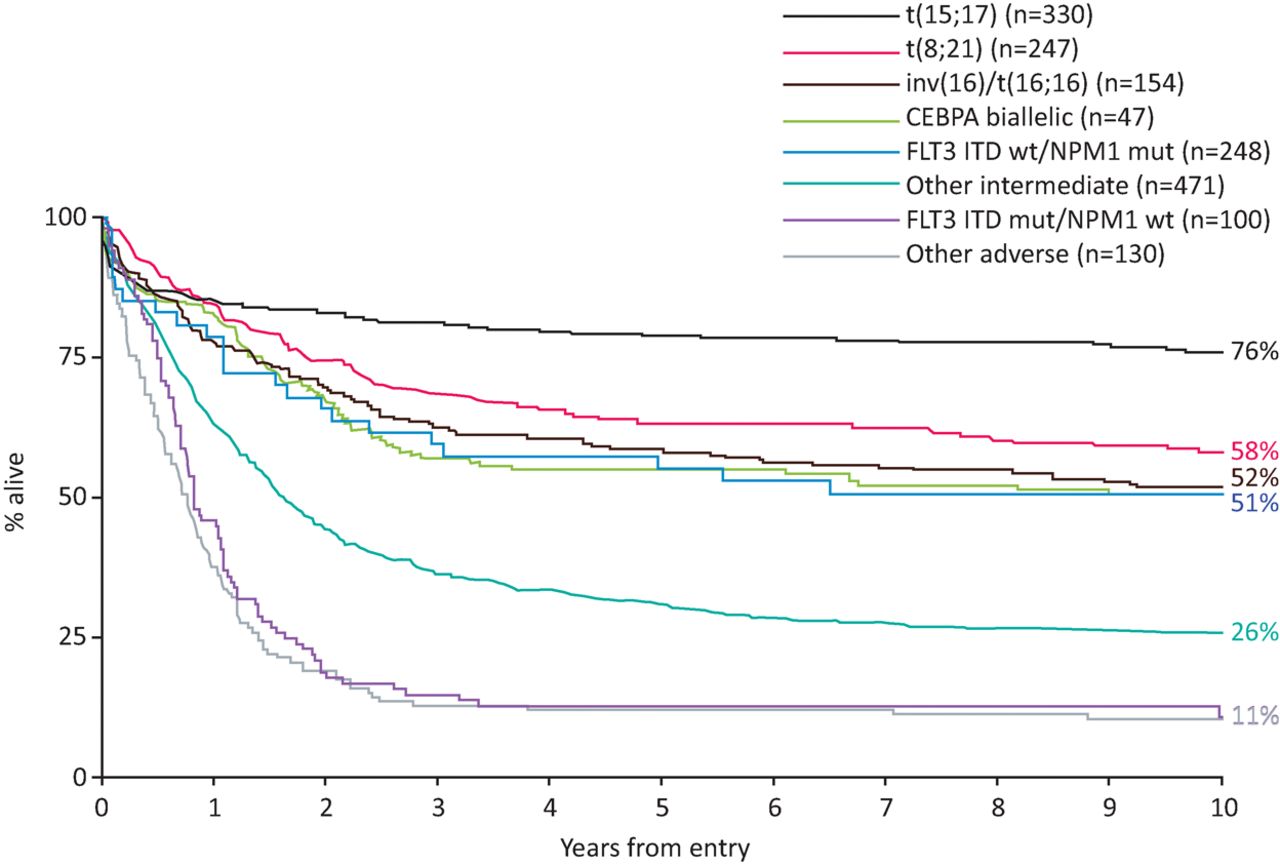
Impact of cytogenetics and CEBPA, NPM1 and FLT3-ITD mutations on patient outcomes. Overall survival is shown for younger adults (age 16–59 years) with AML treated in the MRC AML10 and AML12 trials, according to cytogenetic and molecular abnormalities as per the European LeukemiaNet 2010 risk stratification.3 AML = acute myeloid leukaemia; ITD = internal tandem duplication. Reproduced with permission from Smith et al.4
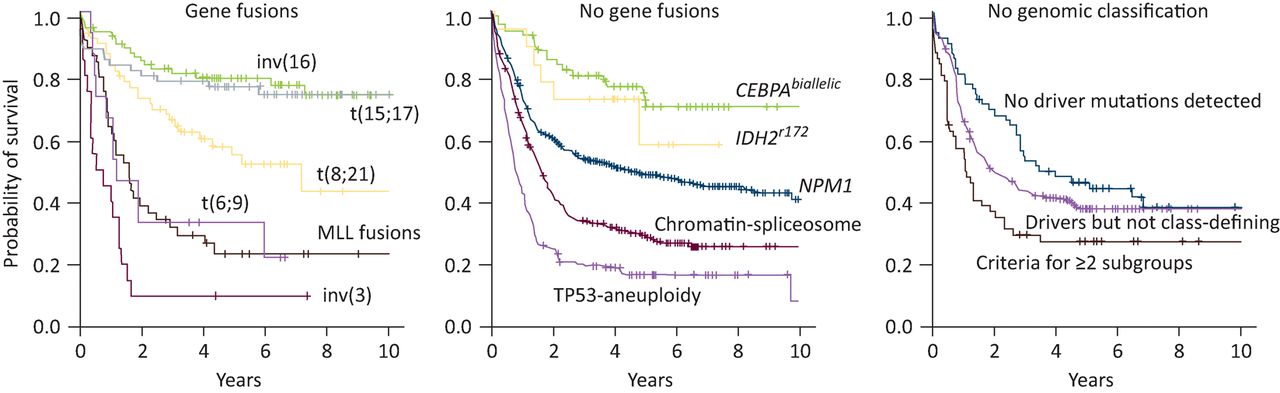
The advent of fast and affordable next-generation sequencing technologies has led to the discovery of many novel somatic mutations in AML.9–11 Systematic studies have shown that AML is genetically less complex than many solid cancers, with a median of approximately 13 somatic mutations per AML genome, of which five are in recurrently mutated genes.10,12 On average, patients have 1–2 distinct leukaemic subclones in addition to the founding clone, although this is almost certainly an underestimate.10 Distinct patterns of clonal evolution emerge; mutations in genes encoding epigenetic regulators such as DNMT3A, TET2 and ASXL1 tend to be founder events that give rise to a pre-leukaemic clone, whereas mutations in signalling pathways and transcription factors occur later in leukaemic transformation.12,13 Recently, an analysis of leukaemia-associated genes on a large patient cohort identified new interactions between genetic mutations.11 Co-mutation patterns classify AML into 11 distinct clinical subgroups with different outcomes (Fig 2). Although this classification may be refined in the years to come, it represents a major step forwards. A similar trend towards extensive molecular profiling is occurring in other haematological malignancies, particularly in acute lymphoblastic leukaemia14 and chronic lymphocytic leukaemia.15,16
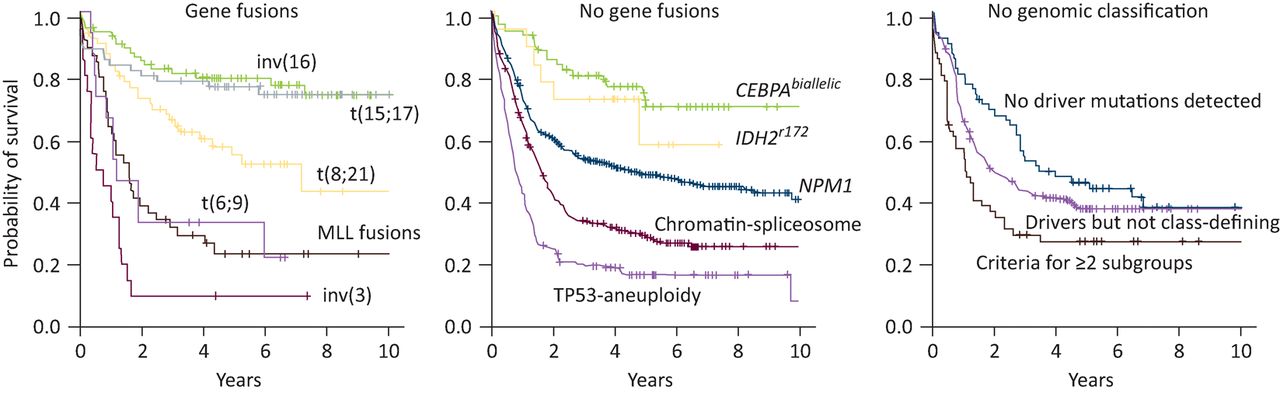
Kaplan-Meier curves showing overall survival among AML patients in the 11 genomically defined subgroups and those who did not have a straightforward classification, as defined by Papaemmanuil et al.11 Note the central panel represents patients with a normal karyotype who would all previously have been classified as intermediate-risk, with the exception of those with NPM1 and CEBPA biallelic mutations. AML = acute myeloid leukaemia. Reproduced with permission from Papaemmanuil et al.11
Targeting signalling pathways
The development of imatinib in the 1990s prompted trials of tyrosine kinase inhibitors in many other cancers. Growth-promoting signalling pathways are aberrantly activated in many haematological malignancies. One such target is Fms-related tyrosine kinase 3 (FLT3), which is expressed on haematopoietic progenitor cells and on most leukaemic myeloblasts. FLT3-ITD mutations occur in approximately 30% of AML and confer a poor prognosis because of high relapse rates.7,9
First-generation FLT3 tyrosine kinase inhibitors have suffered from significant off-target effects and toxicity, and their use as single agents in relapsed or refractory disease has failed to produce sustainable responses.17 Second-generation inhibitors are much more selective for FLT3 and have shown better response rates in this setting. These agents are effective in killing leukaemic blasts, but most patients have incomplete blood count recovery and relapse. This may be because suppressing FLT3-mutant cells does not eliminate the founding leukaemic clone and resistance occurs because of secondary mutations in the FLT3-tyrosine kinase domain.17 The relatively short duration of response means that these agents may be useful in bridging young patients to an allo-HSCT, as demonstrated by a recent phase III study using the multikinase inhibitor midostaurin in combination with induction chemotherapy.18
Numerous kinase inhibitors are in development targeting the KIT, JAK/STAT and B-cell receptor signalling pathways among others.19–21 Resistance invariably develops with single-agent therapy; this is often associated with emergence of resistant subclones with mutations affecting the target protein or in downstream effectors.17,19 It is therefore likely that to achieve lasting responses kinase inhibitors will need to be used in combination with other therapies.
Differentiation therapy
In the transformation to acute leukaemia, malignant haematopoietic cells acquire a block in differentiation, which results in impaired production of mature blood cells and accumulation of immature progenitor cells in the bone marrow. In the 1970s and 1980s, the concept arose that it might be possible to treat leukaemia by inducing differentiation of leukaemic cells. This led to the discovery that all-trans retinoic acid (ATRA) was effective in treating acute promyelocytic leukaemia (APL). In APL, a translocation between chromosomes 15 and 17 results in a fusion between the promyelocytic leukaemia (PML) gene and retinoic acid receptor-alpha (RARα). The PML-RARα fusion oncoprotein represses transcriptional expression of target genes essential for granulocytic differentiation. ATRA leads to degradation of the fusion protein, resulting in cell maturation and resolution of coagulopathy. The first APL patients were treated with ATRA in Shanghai, China in 1985, achieving complete remission in 96% of patients.22 Subsequent trials showed that by combining ATRA with low-dose chemotherapy, long-lasting remissions could be achieved. In 1992, it was reported that the traditional Chinese medicine Ailing-1, which contains high levels of arsenic trioxide, induced remission in APL patients who had relapsed after treatment with ATRA. Since then, therapy for APL has been refined such that a chemotherapy-free regimen combining ATRA and arsenic trioxide is likely to become the standard of care for low-risk disease.23
Over the last 30 years, these developments have transformed APL from a fatal to disease to one that is essentially curable, but unfortunately treatment with ATRA and arsenic has not been successful in non-APL AML. Recently, isocitrate dehydrogenase inhibitors have emerged as a new class of differentiation therapy. 15–20% of myeloid leukaemias and most gliomas have mutations affecting the isocitrate dehydrogenase enzymes, IDH1 and IDH2.10 IDH enzymes catalyse the conversion of isocitrate to alpha-ketoglutarate (α-KG), which constitutes a key step in the citric acid cycle. Several isoforms exist: IDH1 is localised to the cytoplasm, whereas IDH2 is mitochondrial. Mutations in IDH1/2 result in neomorphic enzyme activity converting α-KG to the oncometabolite 2-hydroxyglutarate (2-HG). By inhibiting the activity of histone and DNA demethylases, 2-HG alters gene expression and causes a differentiation arrest.
Small molecule inhibitors of mutant IDH1 and IDH2 are currently in phase I/II clinical trials. Interim results for AG-221 (Enasidenib), an orally available inhibitor of mutant IDH2, have been reported in AML and myelodysplastic syndrome.24,25 Of 138 patients with relapsed/refractory AML, most of whom had high-risk, heavily pretreated disease, 41% showed a response, with complete remission in 23%. In myelodysplastic syndrome patients, the overall response rate was 53%.25 Treatment induces differentiation of leukaemic blasts into mature myeloid cells and, interestingly, many patients had prolonged stable disease with recovery of neutrophil and platelet counts despite persistence of leukaemic blasts in the bone marrow. It is notable, however, that in contrast to preclinical animal studies, not all patients respond and none are cured with single agent therapy, which illustrates that the complexity of human disease is not fully captured in current preclinical models.
Immunotherapy
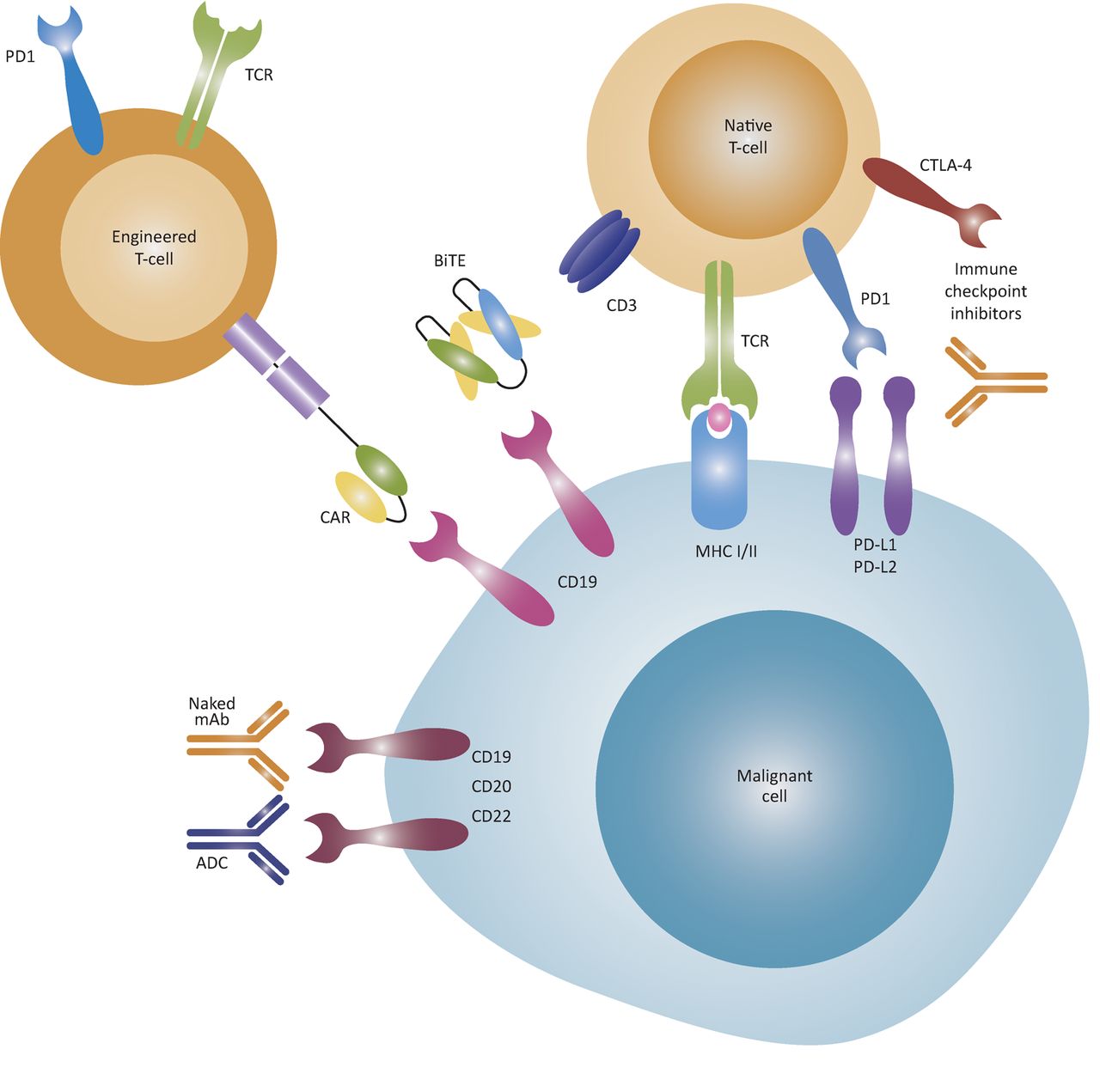
Allo-HSCT, in which a patient is infused with bone marrow stem cells from a genetically similar but not identical donor, remains the most effective long-term treatment for AML. In patients who are fit for this highly intensive treatment, it leads to cure in 50–60%.26 Recognition that the graft-versus-leukaemia effect was critical in mediating cure following allo-HSCT naturally led to an interest in achieving curative anti-cancer immune responses while avoiding graft-versus-host disease. While numerous strategies to improve cure rates after allo-HSCT have been trialled, benefits gained from boosting the graft-versus-leukaemia effect are largely counterbalanced by increases in morbidity and mortality from graft-versus-host disease. Recently, novel immunotherapies have emerged, including antibody-drug conjugates, bispecific T-cell engaging antibodies, immune-checkpoint inhibitors and adoptive T-cell therapy (Fig 3).
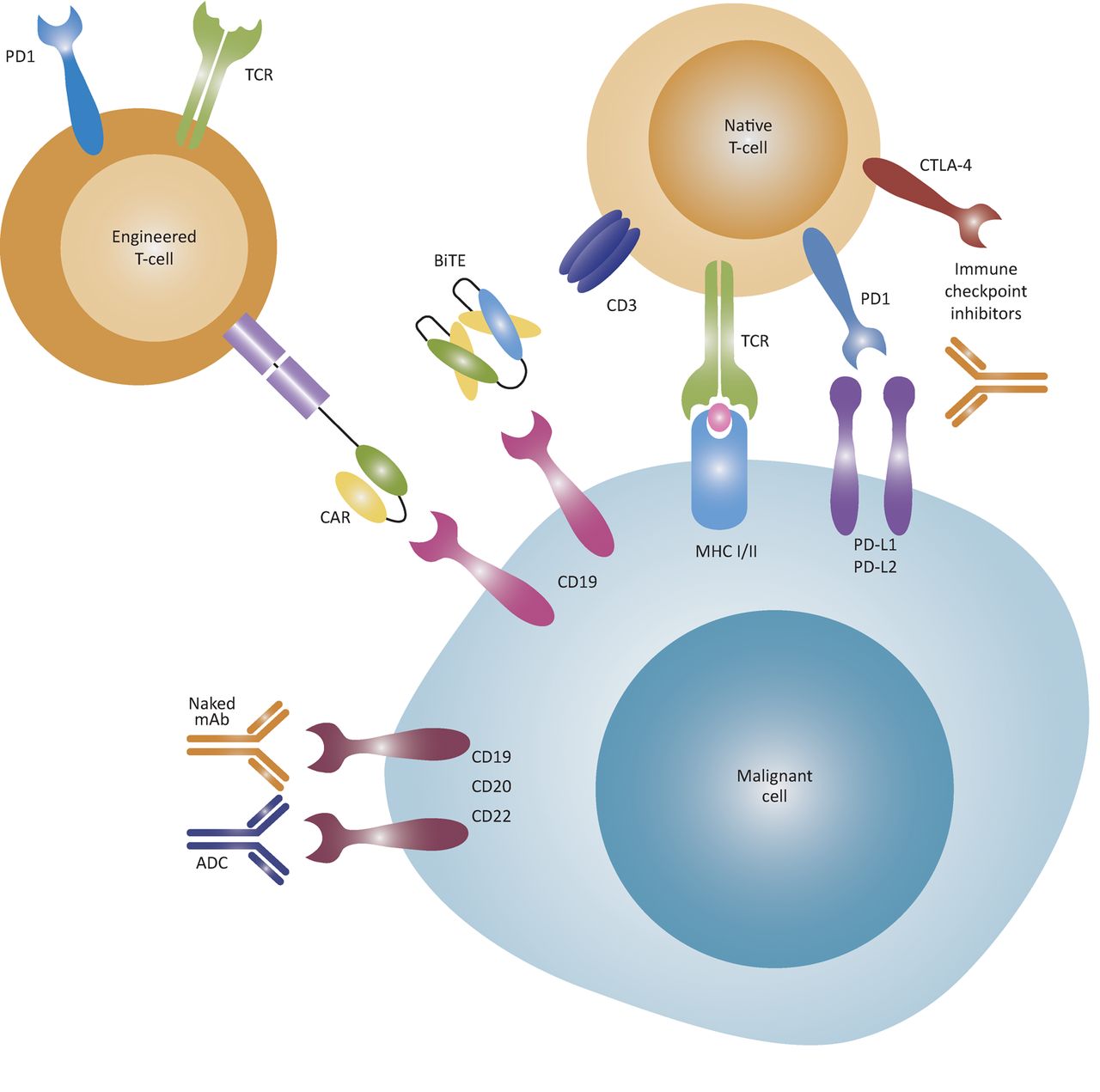
Mechanisms of action of immunotherapies. ADCs are monoclonal antibodies linked to an effector agent, such as a cytotoxic drug, allowing the drug to be targeted to malignant cells. BiTE antibodies bring T-cells and malignant cells into close proximity through dual antigen binding, such as to the CD19 B cell antigen and the CD3 T-cell co-receptor, and can induce T-cell activation without co-stimulatory signals. Immune-checkpoint inhibitors block the inhibitory PD1 and CTLA-4 co-receptors on T-cells to avert T-cells from anergy. T-cells can be engineered to express CARs that recognise cell-surface molecules independent of MHC. Later-generation CARs have both T-cell receptor and co-stimulatory signalling components, thereby activating the T-cells without additional co-stimulatory signal. Alternatively, T-cells can be engineered to express tumour-specific T-cell receptors that recognise intracellular tumour antigens presented by MHC molecules. ADC = antibody-drug conjugate; BiTE = bispecific T-cell engaging antibody; CAR = chimeric antigen receptor; CTLA-4 = cytotoxic T-lymphocyte-associated protein 4; mAb = monoclonal antibody; MHC = major histocompatibility complex; PD1 = programmed cell death protein 1; PD-L1 = programmed cell death 1 ligand 1; TCR = T-cell receptor. Reproduced from Batlevi et al29 with permission from Macmillan Publishers Ltd.
Antibody-drug conjugates
Antibody-drug conjugates target an effector agent, such as a cytotoxic drug, to cancer cells by linking it to a monoclonal antibody.27 CD33 is an attractive target in myeloid malignancies because it is expressed on >90% of myeloblasts (as well as normal myeloid cells). Gemtuzumab ozogamicin, which combines a humanised anti-CD33 antibody with the highly potent cytotoxic agent calicheamicin, received accelerated US Food and Drug Administration (FDA) approval for treatment of AML in 2000, but was subsequently withdrawn because of deaths from haematological and liver toxicity.27 However, a modified dosing schedule allows the drug to be given safely with induction chemotherapy and this increases survival.28 Brentuximab vedotin, an anti-CD30 antibody linked to the microtubule-inhibitor monomethyl auristatin E, has been approved for treatment of Hodgkin lymphoma and systemic anaplastic large-cell lymphoma; numerous other antibody-drug conjugates are in development.27 Evidently, the choice of antigen target is of key importance and determines the side effect and toxicity profile, as illustrated by myelosuppression caused by gemtuzumab ozagomicin. The lack of differentially expressed proteins makes this particularly challenging in myeloid leukaemia.
Bispecific T-cell engaging antibodies
Bispecific T-cell engaging antibodies (BiTEs) are engineered fusion proteins comprised of single chain variable fragments (scFv) of two distinct antibodies linked by a short peptide.29 One variable fragment binds the CD3 T-cell co-receptor and the second binds a protein on the target cell. Thus, in contrast to conventional antibodies, BiTEs redirect host T-cells to cancer cells leading to sustained T-cell activation, proliferation and cancer-directed cytotoxicity. The first BiTE to be developed for AML is the CD33/CD3 construct AMG 330, which has demonstrated encouraging preclinical activity and recently entered phase I clinical trials.30 In B-cell acute lymphoblastic leukaemia and non-Hodgkin lymphoma, blinatumomab, which is targeted to the B-cell antigen CD19, has shown efficacy even in relapsed/refractory patients with high tumour burden who have few treatment options left.31,32 Most adverse effects are mild to moderate, but neurological toxicities – including confusion, tremor, focal neurologic deficits and seizures – as well as transient B-cell depletion and neutropenia have been reported.
Immune-checkpoint inhibitors
The interaction between a T-cell and an antigen presenting cell through the T-cell receptor antigen / major histocompatibility complex involves simultaneous co-stimulatory and co-inhibitory signals. The balance of these signals determines whether T-cell activation occurs. The remarkable success in melanoma and other solid tumours of ‘immune-checkpoints inhibitors’ targeting CTLA-4 and PD-1 has prompted trials in haematological malignancies. Early-phase studies are underway in myeloid malignancies, with durable responses observed in a trial of the CTLA-4 antibody ipilimumab in patients that had relapsed post allo-HSCT.33 This drug class is particularly attractive in lymphoid malignancies, where PD-1 ligands are frequently overexpressed and there is evidence of a present but ineffective anti-tumour immune response, most obviously in Hodgkin lymphoma. Indeed, the PD-1 inhibitor nivolumab has shown excellent results in Hodgkin lymphoma,34 leading to accelerated FDA approval last year. Toxicity profiles are mostly immune-related, including colitis, pneumonitis, neutropenia, hyperthyroidism and rash.
A novel innate immune checkpoint has been identified in CD47, which acts as a regulator of phagocytosis by binding the inhibitory SIRPα receptor on macrophages and other phagocytes.35 CD47 is upregulated on leukaemia stem cells, which are resistant to chemotherapy and thought to cause disease relapse. A humanised anti-CD47 antibody recently entered phase I clinical trials in patients with AML and solid tumours.35
Adoptive T-cell therapy
Adoptive T-cell transfer involves ex vivo selection and expansion of autologous T-cells before reinfusion into the patient. The ability to genetically engineer T-cells with a chimeric antigen receptor (CAR) or modified T-cell receptor targeted to a cancer-specific antigen radically enhances the potential of this platform.
CARs consist of a scFv targeted to a cell-surface tumour antigen, linked to the activating intracellular-signalling domain of the T-cell receptor. CAR T-cells recognise the target antigen through the antibody domain, resulting in T-cell-mediated killing independent of major histocompatibility complex-antigen presentation. CARs have undergone several generations of development to incorporate co-stimulatory domains that amplify the activating signal and endow the transduced T-cell with potent cytotoxic activity.29 In 2014, a landmark trial at the University of Pennsylvania achieved complete remission in 27 of 30 children with relapsed/refractory acute lymphoblastic leukaemia using CD19 CAR T-cells.36 Since then, there have been positive results in chronic lymphocytic leukaemia and B-cell lymphoma with frequent long-lasting remissions,29 and phase I clinical trials have started in AML.37 However, the use of CAR T-cell therapy in AML is limited by a lack of any truly leukaemia-specific cell-surface antigens that are not expressed by normal haematopoietic stem and progenitor cells.37 Long-term persistence of CAR T-cells directed against myeloid antigens would therefore be likely to eliminate normal haematopoiesis.
In contrast, T-cells engineered to express a tumour-specific T-cell receptor can recognise intracellular tumour antigens presented by human leukocyte antigen molecules, giving access to a much larger range of targets for which antibodies are unsuitable. In an early trial investigating this approach, adoptive transfer of T-cells expressing an affinity-enhanced T-cell receptor specific to the NY-ESO-1 cancer testis antigen achieved a complete or near-complete response in 14/20 patients with advanced multiple myeloma.38
Adoptive T-cell therapy represents the epitome of personalised cancer medicine; it is expensive, requires specialised facilities and challenges need to be addressed before it can be adopted widely in clinical practice. However, the ability to achieve potent anti-tumour immune responses while avoiding the debilitating graft-versus-host disease associated with allogeneic transplant, gives this approach great promise. Critically, in contrast to small molecule inhibitors, both T-cell therapy and other immunotherapies have the potential to produce durable responses.
Conclusions
Advances in understanding the genomic landscape of haematological malignancies over the last decade mean that it is now possible to precisely define the disease in an individual patient. This makes it a priority to establish rapid molecular diagnostics in the clinical setting so that results can influence treatment decisions in real time.19 There is much speculation on the possibilities of personalised therapy; while current therapeutic strategies are based on genetic analysis to some degree, treatment categories remain rather broad because there are few first-line treatment options and knowledge of how particular genetic mutations affect treatment response is incomplete.
Navigating the vast array of novel drugs will be a considerable challenge for haematologists in the years to come. We are yet to learn in which settings each class is most effective, which combinations are safe, the optimal dosing schedules, and how to manage the toxicity and side effect profiles of each. Combination therapy will be needed to achieve the greatest benefit from molecularly targeted treatments. For example, it is plausible that targeted inhibitors (eg FLT3 and IDH inhibitors) could be combined with induction chemotherapy as first-line treatment for AML, while immunotherapies may prove effective in eradicating minimal residual disease and perhaps eventually as an alternative to allogeneic stem cell transplant. Similarly, in lymphoid diseases immuno-oncology agents may replace consolidation therapies, such as chemotherapy and autologous stem cell transplantation. Thus, there is a need to design clinical trials that effectively and rapidly test combinations of agents. This is particularly challenging because many novel therapies are targeted to specific mutations that are present in only a small subgroup of patients; therefore, recruiting sufficient patients into trials to achieve adequate statistical power is likely to require international collaboration.
Targeted treatments are only beginning to demonstrate durable benefits, but it is clear that in the near future we are likely to see a radically different therapeutic landscape after years where there was little change. For haematologists, GPs and physicians who provide care for patients with haematological malignancies, an awareness of all of these issues will be important as the landscape of treatments evolves.
Genomic technologies used in clinical management of haematological malignancies
Conflicts of interest
The authors have no conflicts of interest to declare.
Author contributions
NAJ wrote the first draft of the manuscript and both authors reviewed and edited the final version.
Acknowledgements
NAJ is supported by NIHR Biomedical Research Centre, based at Oxford University Hospitals Trust, Oxford, the Medical Research Council and Leuka (Grant Ref: MR/R002258/1). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
- © Royal College of Physicians 2018. All rights reserved.
References








{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- ABSTRACT
- Introduction
- From morphological towards genomic classification of disease
- Targeting signalling pathways
- Differentiation therapy
- Immunotherapy
- Antibody-drug conjugates
- Bispecific T-cell engaging antibodies
- Immune-checkpoint inhibitors
- Adoptive T-cell therapy
- Conclusions
- Conflicts of interest
- Author contributions
- Acknowledgements
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.