
ABSTRACT
Alcohol-related liver disease (ARLD) and non-alcoholic fatty liver disease (NAFLD) are leading causes of chronic liver disease globally. Both ARLD and NAFLD are multifactorial and refer to a spectrum of disease severity, ranging from steatosis through steatohepatitis to fibrosis and cirrhosis. Both diseases exhibit substantial inter-patient variation in long-term outcomes and are best considered complex disease traits where genetic and environmental factors interact to mediate disease severity and progression. Here, we briefly review the current literature describing the best validated genetic modifiers that influence severity of these liver conditions, including variants of the genes PNPLA3, TM6SF2 and MBOAT7, which have also been implicated in lipid dysregulation.
Introduction
Alcohol-related liver disease (ARLD) and non-alcoholic fatty liver disease (NAFLD) are leading causes of chronic liver disease globally. Both ARLD and NAFLD are multifactorial and refer to a spectrum of hepatic disorders. The conditions share common histological features, ranging from steatosis (defined as an accumulation of fat in >5% of hepatocytes) to steatohepatitis, fibrosis and cirrhosis. Both diseases display inter-patient differences in disease severity and progression that ultimately lead to substantial differences in patient outcomes. It is well established that excessive alcohol consumption leads to chronic liver disease; however, only a small proportion of those people consuming large amounts of alcohol develop alcoholic steatohepatitis, fibrosis or cirrhosis. A similar trend is observed in NAFLD. Only a small proportion of non-alcoholic steatohepatitis patients will go on to develop more severe, life threatening disease, with 10–30% of patients developing advanced fibrosis or cirrhosis, for which liver transplant is the only treatment currently available.1
The factors that mediate disease progression in both ARLD and NAFLD have been shown to involve a complex interplay between environmental risk factors (eg sustained high levels of alcohol consumption or a sedentary lifestyle and poor diet, features of the metabolic syndrome) and genetic risk factors. To date, this interplay and all the factors involved are incompletely understood.
Genetic modifiers of disease progression
Epidemiological, familial and twin studies have shown familial clustering of NAFLD, with an estimated 50% heritability for both degree of hepatic steatosis and severity of non-alcoholic steatohepatitis-fibrosis having been reported.2 Twin studies carried out in ARLD cohorts also support a genetic contribution to disease risk, with the prevalence of ARLD being nearly threefold higher in monozygotic than dizygotic twins.3 Genome-wide association studies (GWAS) have been utilised to identify genetic risk loci for ARLD and NAFLD.4–6 From these studies, a non-synonymous genetic variant that causes an amino-acid substitution (I148M) in patatin-like phospholipase domain containing-3 (PNPLA3) has emerged as a major risk factor for chronic liver disease progression. More recently, a variant causing an amino acid change (E167K) in another gene, transmembrane 6 superfamily member 2 (TM6SF2), has also been shown to be an important susceptibility modifier for progressive liver disease. Similarly, a variant (rs641738) in the membrane bound O-acyltransferase domain containing 7-transmembrane channel-like 4 (MBOAT7) gene has also garnered substantial interest.7 All three genes have now been associated with both ARLD and NAFLD disease severity.
PNPLA3
A common PNPLA3 non-synonymous variant (rs738409 c.444 C>G p.I148M) has been reported, on several occasions, to be associated with susceptibility to steatosis, steatohepatitis and hepatic inflammation and fibrosis.4,8,9 This association was first reported by Romeo et al in 2008 in a GWAS of non-synonymous single nucleotide polymorphisms in NAFLD. Steatosis was assessed using proton magnetic resonance spectroscopy in a cohort of 2,111 patients in the Dallas Heart Study. PNPLA3 rs738409 was significantly associated with increased hepatic triglyceride content and an increase in hepatic inflammation. In the same study, an association between the variant and increased levels of serum alanine aminotransferase in a Hispanic subpopulation was also observed.4 This association was then extended to progressive steatohepatitis in 2009.8 The observation that the rs738409 variant mediates disease progression has been repeatedly validated through various candidate gene studies.8,10,11 The clinical importance of PNPLA3 has been highlighted by studies in histologically characterised cohorts associating PNPLA3 with disease severity, such as grade of steatohepatitis or severity of fibrosis.10,11
Subsequently, ARLD candidate gene studies have supported PNPLA3 as a modifier of ARLD disease progression. In a systematic review and meta-analysis, carriage of the I148M variant was associated with a 2.1-fold increase in the risk of cirrhosis in ARLD patients that are heterozygous for the variant. This increases to 3.37-fold in ARLD patients that are homozygous for the I148M minor allele.12 The relationship between PNPLA3 rs738409 and progressive ARLD has been further replicated in a European GWAS, with independent validation.7
Further to its association with ARLD and NAFLD, PNPLA3 rs738409 has been reported to be a susceptibility variant for hepatocellular carcinoma (HCC). This significant association has been replicated on a background of NAFLD, ARLD and viral hepatitis.13,14 Strong evidence for the effect of PNPLA3 on HCC comes from Liu et al, who used a two-stage case study to analyse I148M genotypes in HCC populations and the general population and compared the two. They reported the first NAFLD study, conducted on a well characterised European NAFLD cohort, observing that the GG genotype at rs738409 has a 2.3-fold increase in HCC risk in NAFLD patients.13 Future studies are needed to assess the utility of PNPLA3 rs738409 as a biomarker with predictive value in chronic liver disease progression.
Following the confirmed association between PNPLA3 variants and histological disease severity, further work has focused on functional characterisation of rs738409, unpicking the molecular biology of the genetic variant. The C>G mutation causes a isoleucine to methionine change at the amino acid residue level (I148M). In 2010, it was reported in the literature that this change in amino acid sequence hindered the protein’s ability to act as a lipase by disturbing the configuration of the active site. As a result, levels of triglycerides start to accumulate in the liver. Because of this, the I148M mutation has been described as a loss of function mutation.15 It has been shown that PNPLA3 knockout mice showed no significant difference to wildtype mice.16 This was disputed in a recent study, which reported that mice carrying the mutated PNPLA3 protein exhibited a loss of function effect and an accumulation of triglycerides.17 Mouse studies on PNPLA3 have their limitations, as there are variations between species in PNPLA3 gene expression levels. In wildtype mice, the gene is expressed at low levels in the liver when compared with human liver samples.18
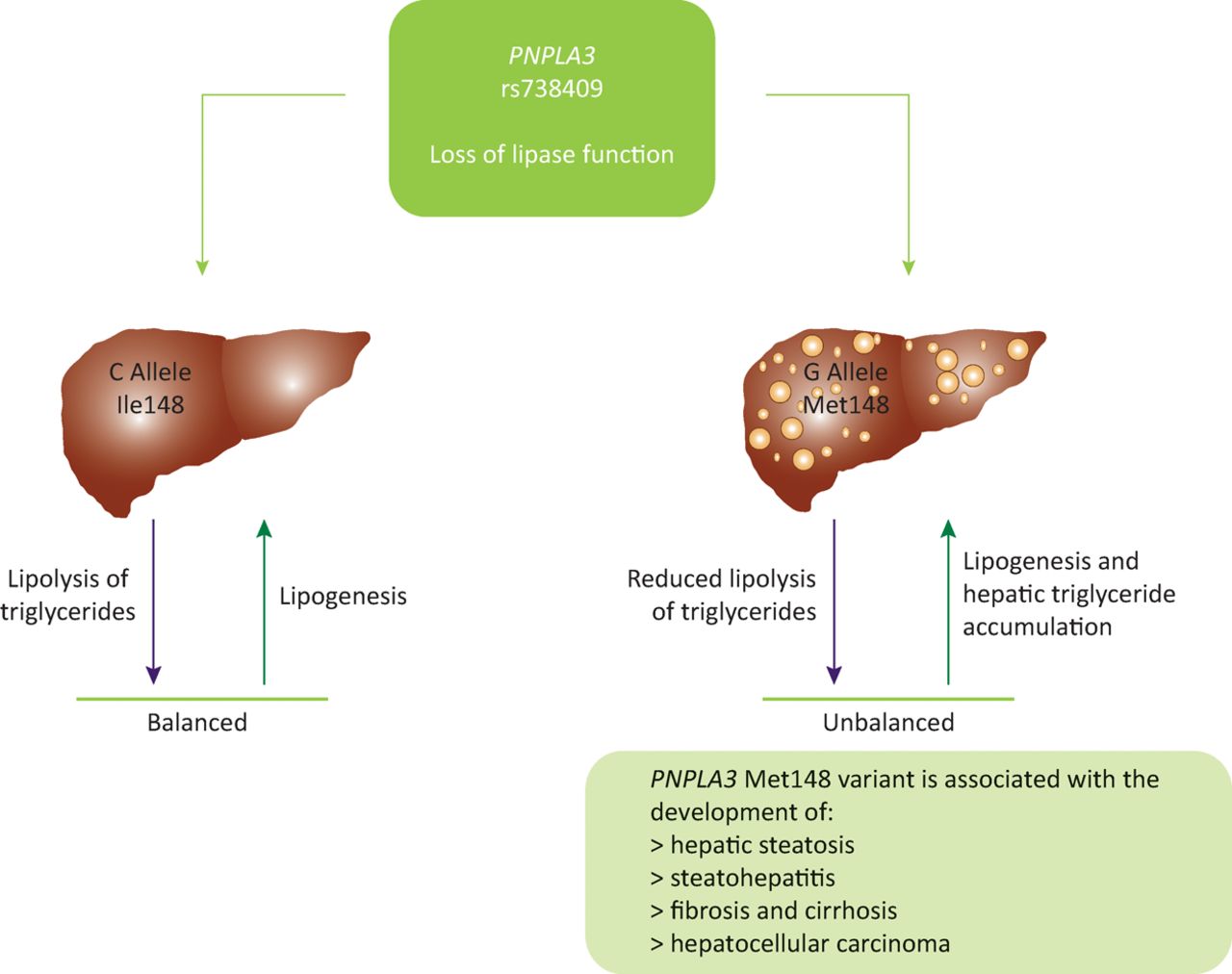
Human studies on the rs738409 variant have shown that PNPLA3 has a role in the regulation of very low density lipoprotein secretion. The variant form reduces the amount of very low density lipoproteins that are secreted.19 As a result of this impaired lipid secretion, lipids accumulate in hepatocytes, leading to the steatosis observed in carriers of the G allele (Fig 1). Further studies to fully determine the pathophysiological function of PNPLA3 are ongoing.
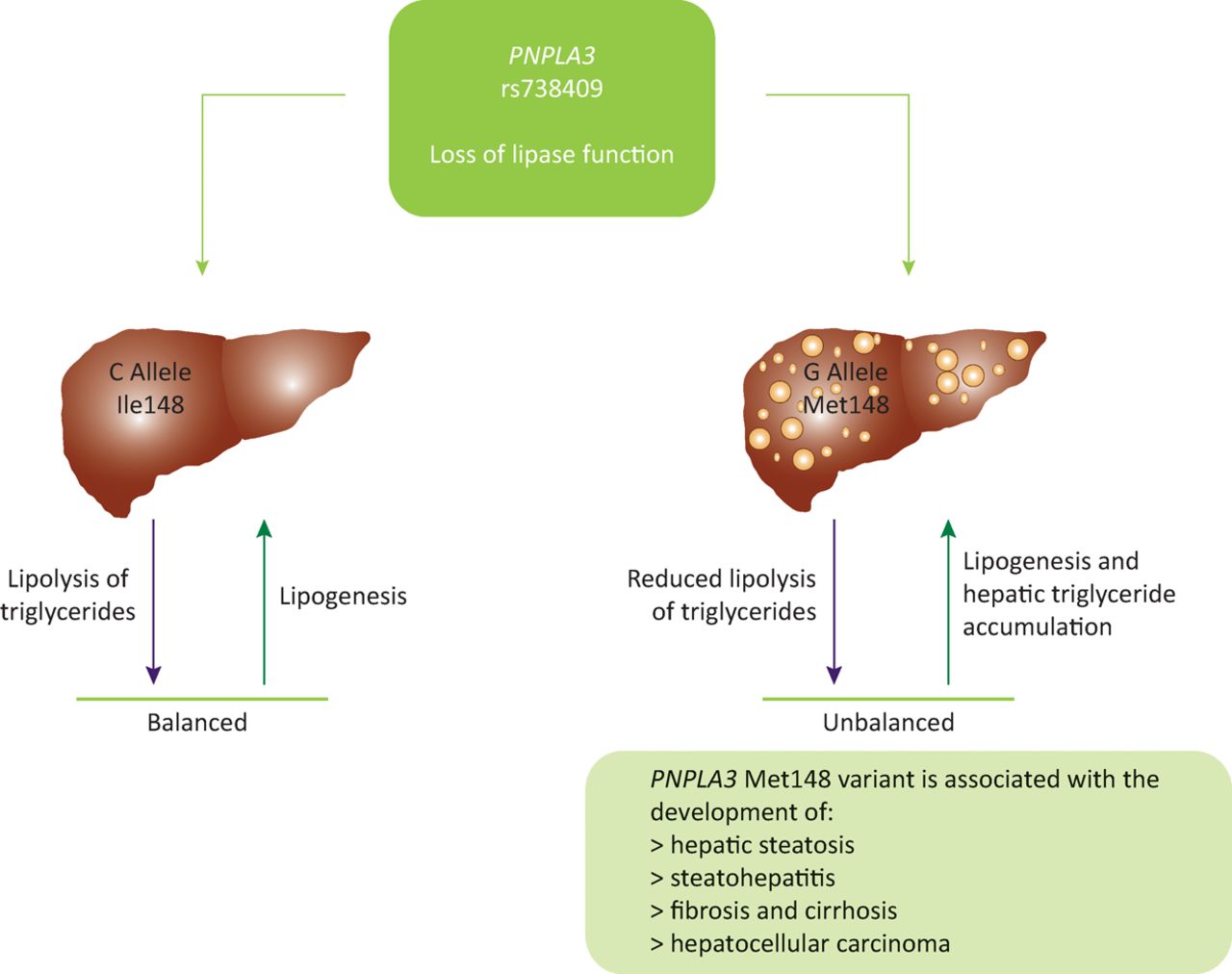
PNPLA3 (rs738409) has a role in the regulation of very low density lipoprotein levels. Wildtype carriers maintain a balance between lipolysis and lipogenesis. Carriage of the mutant G allele results in a loss of lipase function, reducing levels of lipoloysis. This creates an imbalance between lipolysis and lipogenesis, resulting in lipid accumulation in the hepatocytes, which correlates with the increased levels of steatosis observed in carriers of the G allele for rs738409, indicating a possible mechanism for PNPLA3 I148Ms role in the development of steatosis. Ile148 = isoleucine at residue number 148; Met148 = methionine at residue number 148
TM6SF2
The search for genetic modifiers of chronic liver disease progression has since been extended (Table 1)6,20–25 with an exome-wide association study and a second candidate gene study identifying the TM6SF2 (rs5854296 c.449 C>T, p.E167K) as a modifier of hepatic triglyceride content and serum lipoprotein levels as well as a prognostic marker for advanced fibrosis and cirrhosis.6,20 Kozlitina et al carried out the initial exome-wide association study associating rs5854296 [T] with increased hepatic steatosis in their cohort. They also reported an association with degree of liver injury and also decreased levels of plasma lipid, namely low density lipoprotein cholesterol. This association remained significant when conditioned for PNPLA3 rs738409 genotype, indicating that the E167K variant’s effect on steatosis is independent of PNPLA3 status. This association with increased hepatic triglyceride content and decreased levels of serum cholesterol has since been replicated in multiple studies, across multiple ethnicities, in both adult and peadiatic populations.20,22,26
Key studies on the association of the TM6SF2 Glu167Lys variant and NAFLD in adult populations
A validation study conducted in an independent, histologically well-characterised NAFLD cohort reported an association between the E167K variant and the stage and severity of fibrosis (p=6.36×10–6). This association was seen in both a discovery cohort and a validation cohort and remained significant following conditioning for PNPLA3 (p=0.0074; p=0.014). Furthermore, the study subdivided the NAFLD cohort into those presenting mild fibrosis (F0–1) and those with more advanced fibrosis (F2–4). The rs58542926 minor T allele was significantly associated with an increased risk of developing advanced fibrosis (p=1.63×10–5). Carriage of the E167K variant was associated with NAFLD and, in particular, an increased risk of developing advanced fibrosis and cirrhosis.20 Subsequently, an association between the variant and liver fibrosis has also been reported in a paediatric population.26
However, this association has not been validated in every study. Two smaller studies failed to replicate the association with disease severity, reporting that they only observed a weak association of with hepatic steatosis (p=0.038) in univariate analysis.22,23 However, at odds with the findings of Liu et al, they reported no association with fibrosis or degree of fibrosis. This inconsistency is likely due to the smaller studies lacking adequate statistical power because of the low minor allele frequency of the variant and the very limited cohort sizes. Based on the overall consensus of the published literature, TM6SF2 is associated with the severity of NAFLD and a more progressive liver fibrosis phenotype.
Following on from the identification of TM6SF2 as a modifier of NAFLD, a candidate gene study has highlighted an association between the same variant and a risk of ARLD and cirrhosis, with carriers of the T allele having a 1.9-fold increased risk of disease.27 Furthermore, an ARLD GWAS reported that carriers of the E167K variant had a 1.4-fold increased risk of ARLD associated cirrhosis.7
Contributions to the literature regarding the role and function of TM6SF2 have been slow with the gene function remaining elusive. In 2014, Mehdessian and colleagues used confocal microscopy in hepatoma cell lines to determine the subcellular localisation of TM6SF2, determining that TM6SF2 localises to the endoplasmic reticulum (ER) and the ER-Golgi intermediate compartment.28In vitro TM6SF2 knock-down experiments have concluded that knock-down results in a reduction of secretion of triglycerides. The converse effect was seen when cell lines transiently overexpressed TM6SF2. Here, a decrease in the levels of cellular triglycerides was observed, supporting a role for TM6SF2 in hepatic steatosis.
Similar results have been validated in several in vivo studies.6,21,29 Selective knock-down of TM6SF2 in the livers of mice increased hepatic steatosis threefold, while also reducing the the circulating levels of cholesterol and plasma triglycerides.6 CRISPR-Cas9 generated TM6SF2 knockout mice replicated earlier findings that knockout mice had decreased plasma levels of total cholesterol.29 They also observed altered levels of gene expression in genes known to be involved in cholesterol metabolism, leading to the hypothesis that TM6SF2 has a role in cholesterol metabolism in mice.
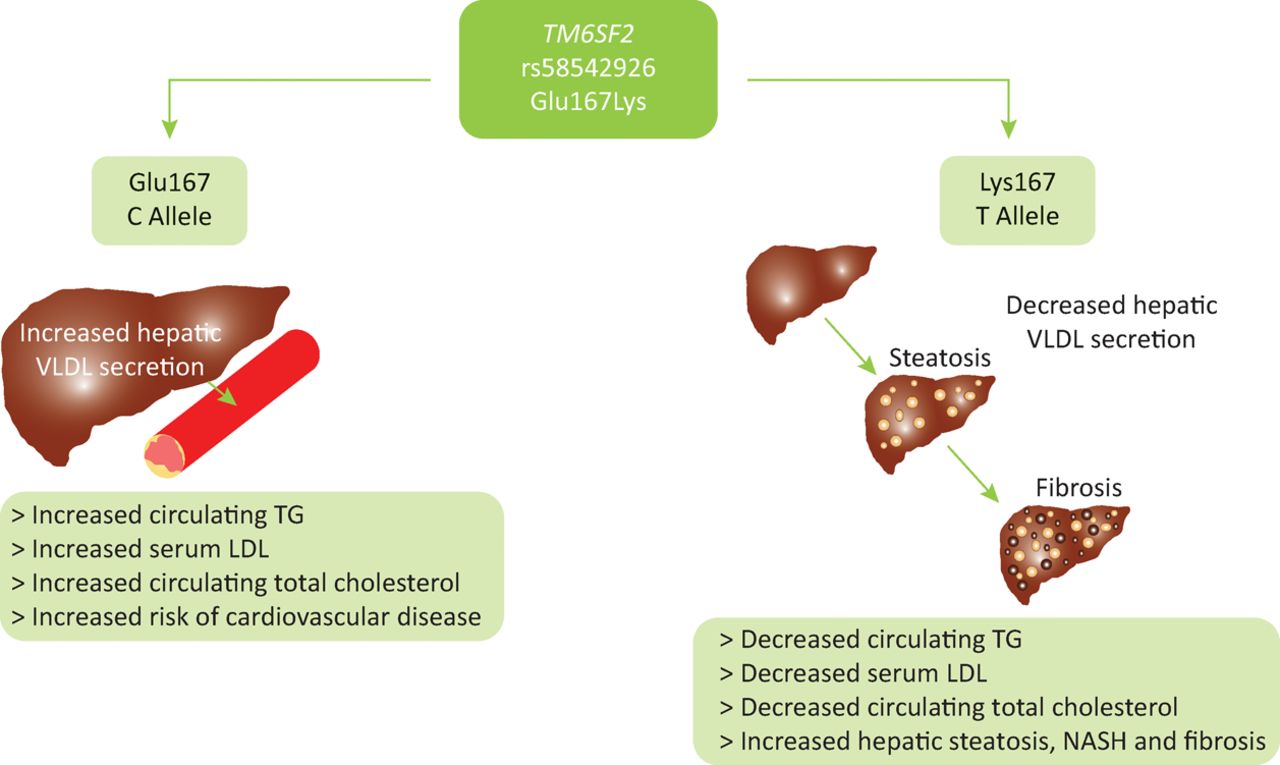
Homozygous carriers of the TM6SF2 E167K variant minor allele have an increased risk of hepatic steatosis, NASH and progression to advanced fibrosis but exhibit lower levels of circulating low density lipoproteins and triglycerides. In contrast, carriage of the wildtype allele promotes secretion of triglycerides and cholesterol into the circulation, protecting the liver at the expense of greater dyslipidaemia. This results in an increased risk of atherosclerosis and cardiovascular disease. Thus, TM6SF2 appears to influence metabolic syndrome-mediated end-organ damage by altering the storage of lipid in the hepatoctyes or its seceretion and effects on the cardiovascular system (Fig 2).30 This highlights the importance of TM6SF2 as a modifier of clinical outcome across multiple organ systems.
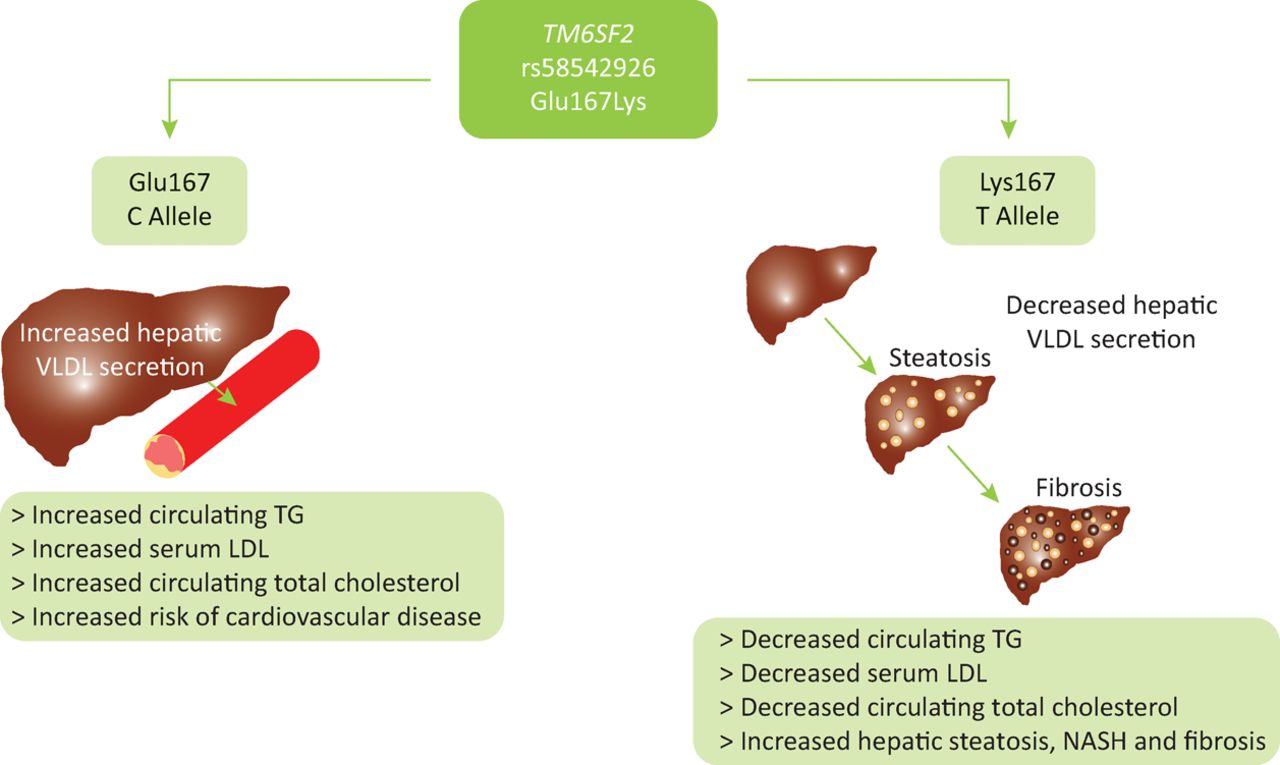
The suggested functional role of the TM6SF2 Glu167Lys variant. Functional studies have indicated that homozygous carriers of the TM6SF2 Glu167Lys variant minor allele have an increased risk of hepatic steatosis and progression to advanced fibrosis, but exhibit lower levels of circulating low density lipoproteins and triglycerides. In contrast, carriage of the wildtype allele promotes secretion of triglycerides and cholesterol into the circulation, protecting the liver, and in turn creating greater dyslipidaemia. This results in an increased risk of atherosclerosis and cardiovascular disease in wildtype carriers. It is suggested that TM6SF2 has a role in very low density lipoprotein (VLDL) secretion and thus acts as a switch between hepatic steatoisis and increased circulating triglycerides. Glu167 = Glutamic acid at residue number 167; LDL = low density lipoprotein; Lys = Lysine at residue number 167; NASH = non-alcoholic steatohepatitis; TG = triglyceride; VLDL = very low density lipoprotein
Other genes
Through various other candidate gene studies, modifiers of alcohol dependence, alcohol metabolism, insulin sensitivity and lipid metabolism have been associated with ARLD and NAFLD.12 The best replicated association with genes involved in alcohol metabolism and ARLD are variants in the alcohol metabolising enzymes alcohol dehydrogenase and acetaldehyde dehydrogenase. Variants in these genes lead to an inability to remove a toxic product of alcohol metabolism – acetaldehyde. Carriers of the variants suffer from an accumulation of acetaldehyde following alcohol consumption, which leads to several unpleasant symptoms that cause them to flush. For this reason, many carriers tend to avoid alcohol consumption, giving them protection from alcohol-associated cirrhosis.31
A 2011 GWAS identified glucokinase regulatory protein (GCKR) as a modifier of NAFLD.5 The rs780094 is in strong linkage disequilibrium with another GCKR SNP (rs1260326) that is known to inhibit GCKR’s ability to respond to fructose-6-phosphate. This inability to regulate glucokinase activity then leads to an increase in glucose uptake in the liver. As a result, levels of malonyl-CoA increase, which promotes hepatic steatosis by acting as a substrate for lipogenesis.32 A further association between GCKR and NAFLD has been established whereby GCKR variants were significantly associated with fibrosis in patients with NAFLD.33
A recent two-step GWAS, carried out in more than 4,000 patients with alcohol-related cirrhosis, identified variants in MBOAT7 (p=9.25×10–10) as a risk loci in ARLD. MBOAT7 is a lysophosphatidylinositol acyltransferase that transfers fatty acids between phospholipids and lysophospholipids.7 Further work is needed to validate the association in ARLD and to understand its role in NAFLD.
Future studies
There are notable similarities in the pathogenesis and genetic modifiers of both ARLD and NAFLD. Study of PNPLA3, TM6SF2 and MBOAT7 highlights these similarities and the key role that aberrant hepatic lipid handling plays in not only NAFLD, but also ARLD. Further multinational studies are underway to validate the genetic modifiers identified in GWAS and to identify other risk loci for chronic liver disease. The challenge remains to examine the functional role of these variants in order to better understand the pathogenesis of the disease and potentially identify new therapeutic targets and strive towards a new era of personalised medicine.
Conflicts of interest
ES is the recipient of an MRC PhD Studentship. ES and QMA are members of the EPoS (Elucidating Pathways of Steatohepatitis) consortium funded by the Horizon 2020 Framework Programme of the European Union under Grant Agreement 634413.
- © Royal College of Physicians 2018. All rights reserved.








{kind=link}
{kind=link}