
ABSTRACT
Inherited metabolic disorders (IMDs) are debilitating inherited diseases, with phenotypic, biochemical and genetic heterogeneity, frequently leading to prolonged diagnostic odysseys. Mitochondrial disorders represent one of the most severe classes of IMDs, wherein defects in >350 genes lead to multi-system disease. Diagnostic rates have improved considerably following the adoption of next-generation sequencing (NGS) technologies, but are still far from perfect. Phenomic annotation is an emerging concept which is being utilised to enhance interpretation of NGS results. To test whether phenomic correlations have utility in mitochondrial disease and IMDs, we created a gene-to-phenotype interaction network with searchable elements, for Leigh syndrome, a frequently observed paediatric mitochondrial disorder. The Leigh Map comprises data on 92 genes and 275 phenotypes standardised in human phenotype ontology terms, with 80% predictive accuracy. This commentary highlights the usefulness of the Leigh Map and similar resources and the challenges associated with integrating phenomic technologies into clinical practice.
Introduction
Inherited metabolic disorders (IMDs) collectively represent one of the most common groups of Mendelian diseases, despite being individually rare (<1:2000) or ultra-rare disorders (<1:2,000,000).1 Many genetic defects underpinning IMDs are enzyme deficiencies within vital biosynthetic and metabolic pathways. However, IMDs are increasingly recognised to include defects of structural proteins, trafficking proteins, chaperones and other regulatory proteins pertaining to a metabolic process.2 IMDs may be associated with debilitating and often fatal phenotypic presentations including epilepsy, intellectual disability, cardiomyopathy, liver failure, myopathy, and immunodeficiency.3,4 A crucial step in the clinical management of IMDs is obtaining a definitive genetic diagnosis, which has clinical, therapeutic, and psychological implications for affected patients and their families (Fig 1). Despite their severity, several IMDs are amenable to clinical intervention such as dietary modification, administration of metabolites, or enzyme replacement therapy, which reduce mortality and morbidity and improve quality of life.4 Unfortunately, patients with IMDs often undergo prolonged diagnostic odysseys because of the complexity of the clinical presentations and the underlying pathobiology typically associated with these disorders. A lack of distinguishing features, non-standardised newborn screening programmes, and a knowledge gap between front-line clinicians and metabolic experts are all obstacles to obtaining specific diagnoses. The genetic heterogeneity of IMDs is another challenging factor in that the most common groups of IMDs may be causally linked to several hundred gene defects. These include mitochondrial disorders, congenital disorders of glycosylation, and lysosomal storage disorders.3,5,6
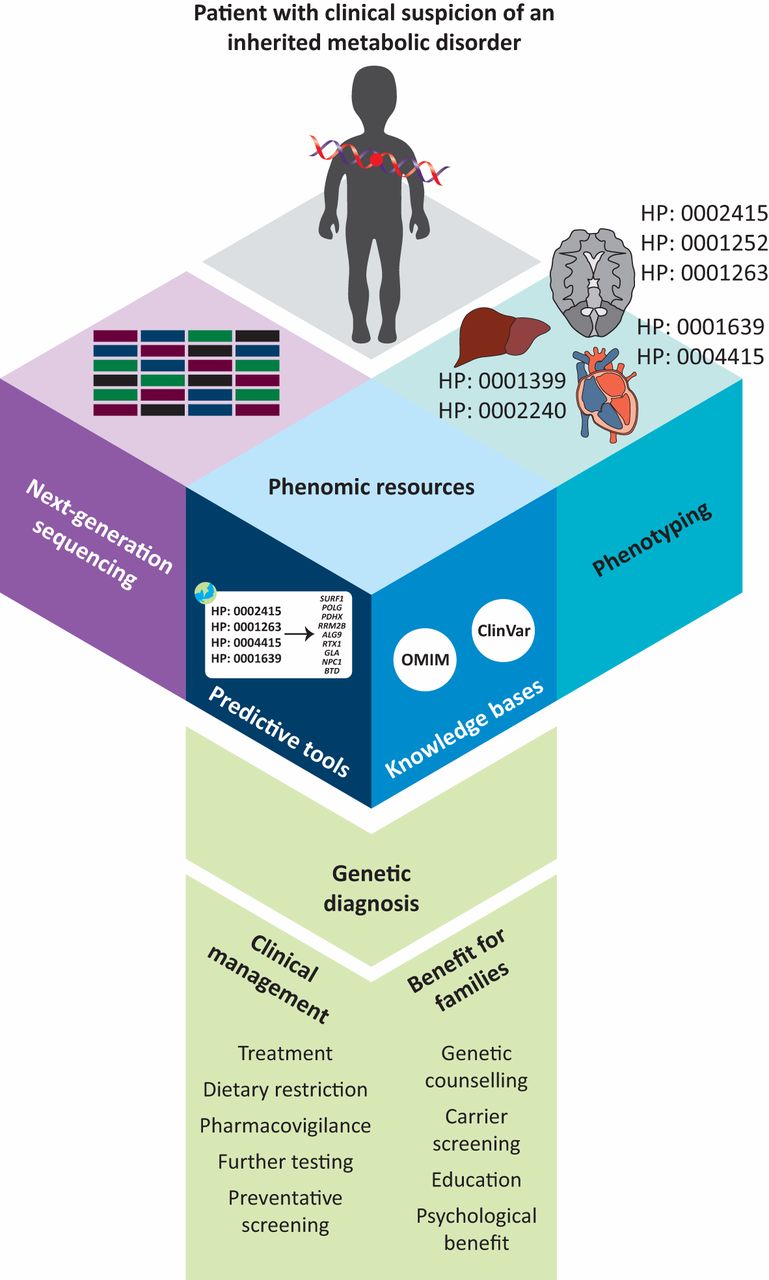
The use of phenomics resources, including predictive tools and rare disease knowledge bases can assist in obtaining a definitive genetic diagnosis when used in conjunction with next-generation sequencing tests such as whole exome or whole genome sequencing. Obtaining a genetic diagnosis improves patient care by providing treatment options, improved pharmacovigilance, and preventative screening for at-risk disorders such as cancer and neurodegenerative diseases. In addition, genetic diagnoses provide benefits to the family by facilitating accurate genetic counselling, as well as providing psychological benefit to parents.
In recent years the use of next-generation sequencing (NGS) technologies has helped to improve the diagnostic rate of IMDs and has exponentially increased the discovery rate of novel causative gene mutations.7 As NGS is increasingly becoming less expensive and more rapid, it is unsurprising that it is emerging as the preferred method of genetic diagnosis in many centres around the world. Furthermore, the use of NGS can in many cases avoid the need for invasive diagnostic tests such as muscle or liver biopsy, limiting the exposure of vulnerable patients, especially children, to general anaesthesia, which in some cases may trigger metabolic decompensation. NGS, however, is not without its challenges, particularly when it comes to prioritising and analysing variants of unknown significance (VUS) or candidate disease genes for potential differential diagnosis. Variant annotation challenges are amplified in those disorders in which there is significant phenotypic heterogeneity. Furthermore, it may be labour intensive and time consuming to comb through knowledge bases or the compendium of literature to determine whether or not a variant is pathogenic. The use of phenomics, that is a complete characterisation of (preferably standardised) phenotypic manifestations associated with a particular gene defect or IMD could have utility in improving the diagnostic rate of difficult-to-diagnose IMDs.8–10 Comprehensive knowledge bases with searchable elements and user-friendly interfaces are cost-effective and potentially efficacious additions to enhance the interpretation of NGS findings and can have secondary benefits as clinical management tools.
Mitochondrial disorders are among the most severe metabolic disorders wherein patients suffer from multi-systemic phenotypes, often resulting in early death. Primary mitochondrial disorders are described as those which primarily or secondarily cause dysfunction of oxidative phosphorylation (OXPHOS) or other elements of mitochondria such as defects in vitamin and cofactor metabolism, aberrant mitochondrial dynamics, and abnormal mitochondrial lipid homeostasis.3 Large-scale NGS gene panel studies currently place the diagnostic rate of mitochondrial disorders between ∼35–60%.11,12 The genetic heterogeneity of mitochondrial disorders and ongoing discovery of novel disease genes are caveats which can deem whole exome sequence (WES) data unsuitable to provide clinicians with enough certainty for a definitive diagnosis in all cases.
We hypothesised that the use of phenomic annotation in mitochondrial disorders could be of diagnostic utility. Mitochondrial disorders are characterised by extreme phenotypic heterogeneity, affecting virtually any organ or system, seemingly in any combination. However, by documenting the phenotypes of reported patients with mitochondrial disease and mapping the associations to causative genes, any emerging patterns can enhance the interpretation of NGS to expedite genetic diagnoses for patients and elucidate novel gene-to-phenotype correlations. To test this strategy, we constructed a novel gene-to-phenotype interaction network using Leigh syndrome as a prototype.
Leigh Map: a case for phenomics-based tools in inherited metabolic disorders
Layout and validation of the Leigh Map
We chose Leigh syndrome as our prototype disorder because it is one of the most common presentations of paediatric mitochondrial disease. Patients frequently die within 2 years of initial presentation.13 Leigh syndrome was initially defined neuropathologically by the presence of bilateral hyperintense lesions in the basal ganglia and/or brainstem.14 Common clinical manifestations include psychomotor retardation, with regression, and progressive neurological abnormalities related to basal ganglia and/or brainstem dysfunction such as movement disorders, speech abnormalities, and defective muscle tone. Furthermore, the majority of patients also present with multisystemic (eg cardiac, hepatic, renal or haematological) phenotypes.13 To date 92 genes are known to cause subtypes of Leigh and Leigh-like syndromes, the majority of which are difficult to distinguish from each other, either clinically or biochemically. We hypothesised that the multisystemic features may help to characterise different genetic subtypes of Leigh syndrome.
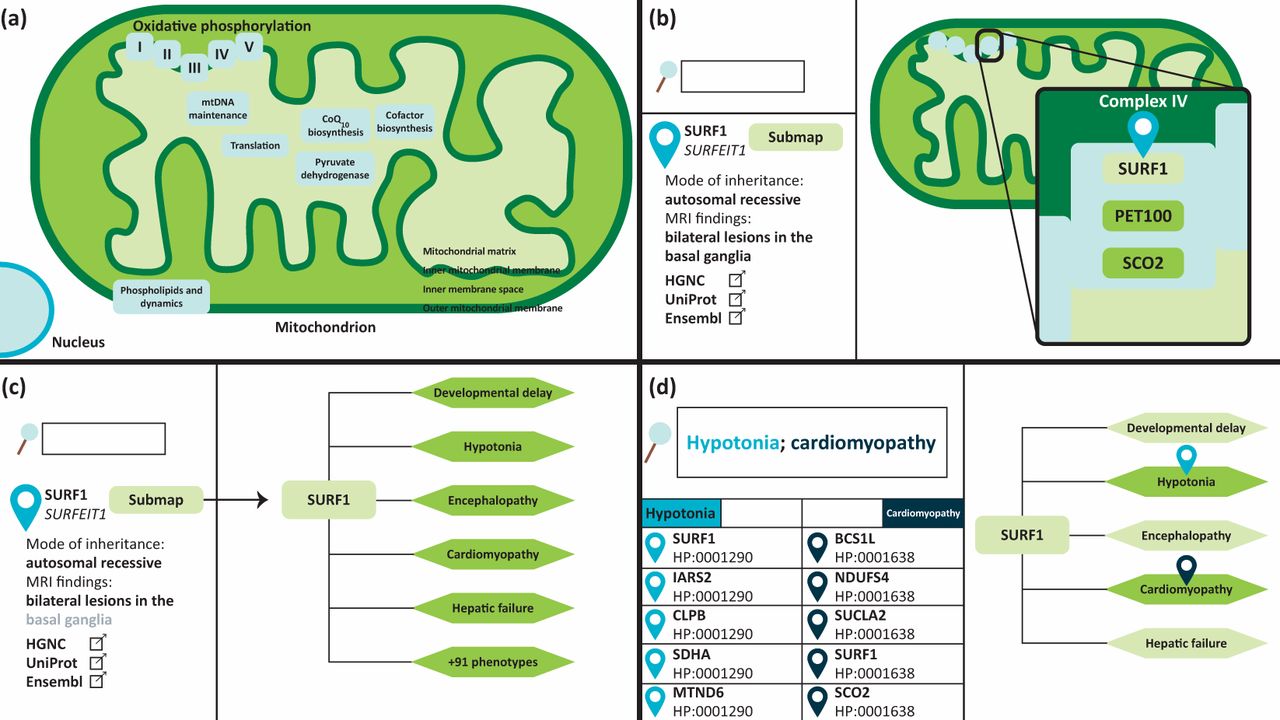
The Leigh Map is a freely available online diagnostic resource for Leigh syndrome (www.vmh.life/#leighmap).10 It is a gene-to-phenotype interaction network encompassing all reported defective genes known to cause Leigh syndrome, and their associated phenotypic abnormalities. The Leigh Map can be utilised to enhance the interpretation of NGS to help prioritise variants, thereby enabling faster and more accurate genetic diagnoses for patients. The network was created by data mining an extensive knowledge base of more than 900 publications to extract genetic and phenotypic data pertaining to Leigh syndrome. In addition, the Leigh Map features additional clinically relevant data. The Leigh Map is built on the Molecular Interaction NEtwoRks VisuAlization (MINERVA) framework, a molecular modelling platform that mimics the user interface of Google Maps to navigate cellular, metabolic, and in our case, phenotypic networks.15 Navigation through the Leigh Map is therefore just as one would navigate Google Maps in that zooming in on compartments reveals progressively more detailed information. To date, 92 causative Leigh syndrome genes are included in the Leigh Map, arranged according to submitochondrial location (inner and outer mitochondrial membranes, intermembrane space, or mitochondrial matrix) and function (OXPHOS, mitochondrial deoxyribonucleic acid maintenance, and mitochondrial dynamics, among others). Clicking on a gene will display relevant information about the gene in the left-hand panel including modes of inheritance, patient demographics, magnetic resonance imaging (MRI) findings, and links to external genetic and scientific databases. Furthermore, each genetic element in the Leigh Map has an accompanying sub-map which documents all phenotypes associated with that particular gene defect. Phenotypes are annotated in human phenotypic ontology (HPO) terms, a system for standardising clinical features that are synonyms.16 For example, ‘decreased muscle tone’ or ‘floppiness’ can be expressed as one HPO term ‘muscular hypotonia’ (HP: 0001252). In total, the Leigh Map currently comprises 236 HPO phenotypes (Fig 2).
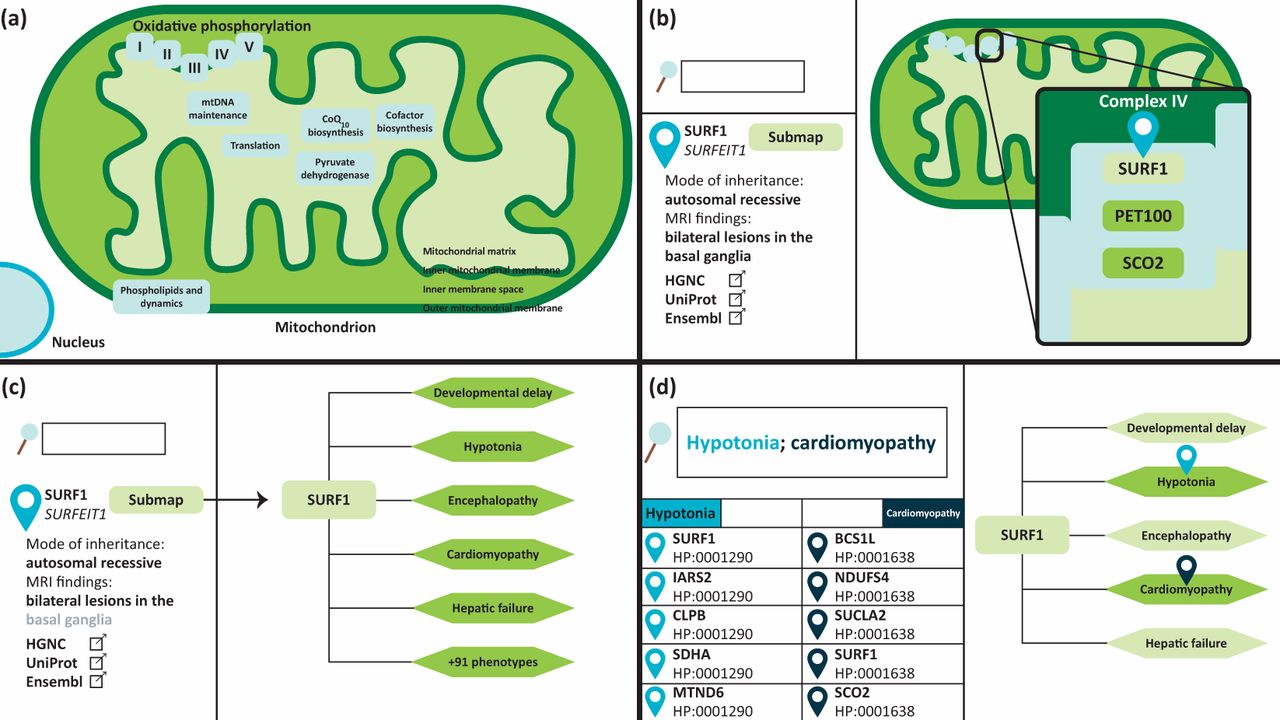
The Leigh Map is an interactive gene-to-phenotype interaction network which can be used as a diagnostic resource for Leigh syndrome. It is built on a platform which mimics the user interface of Google Maps. (a) Causative Leigh syndrome genes are arranged according to their mitochondrial location and function. (b) Zooming in and clicking on a gene will display further information, such as modes of inheritance, MRI findings, patient demographics, and external annotations in the left-hand panel. (c) In addition, each gene has an associated sub-map which displays all phenotypes ever reported in patients with a particular gene defect. (d) The genetic and phenotypic elements of the Leigh map can be queried. Multiple phenotypes can be queried when separated by a semicolon. Results will be displayed on the left-hand panel and clicking on the sub-map of a query result will provide the user with a visual representation of the query phenotypes.
Content query is another Google Maps feature which is embedded within MINERVA. The Leigh Map features more than 1700 gene-to-phenotype interactions, all of which can be queried by the user using the search function. Querying the name or HPO number of a particular phenotype will result in a list of genes with which that phenotype has been associated. The Leigh Map supports the query of multiple phenotypes simultaneously, therefore this feature can be used in a diagnostic setting to elucidate causative disease genes. Conversely, genes can also be queried to browse phenotypes, which can be used as a clinical surveillance tool.10
Two non-clinical independent testers validated the Leigh Map, using unpublished test cases consisting of clinical and biochemical vignettes (excluding genetic information) of patients from a national mitochondrial disease clinic who had a prior established genetic diagnosis of Leigh syndrome. The testers were able to elucidate the correct gene as a candidate in 80% of the test cases. We defined a gene as a candidate if it contained >50% of the query phenotypes. Candidate gene lists typically comprised 8–10 Leigh Map genes, thereby eliminating 85–90% of the genes in the network. The failure to deduce the correct candidate genes for a minority of our test cases was due to the predominant presence of common Leigh syndrome phenotypes such as hypotonia, lactic acidosis, and developmental delay without more specific organ involvement. We found more success in ‘diagnosing’ patients with discriminating phenotypes, usually involving more than one system including cardiomyopathy, hepatopathy, and renal tubulopathy.
Limitations and future prospects
Given its success rate in predicting causative genes by non-clinical individuals, the Leigh Map is an efficacious diagnostic resource that, in combination with WES or whole genome sequence (WGS) data and metabolic testing in blood, urine and/or cerebrospinal fluid, can be used by clinicians to provide patients with accurate diagnoses or to guide further biochemical investigation.
Currently the most significant limitation of the Leigh Map is the lack of a multiple advanced search facility. Future work aims to implement this feature to the network, in addition to a scoring system so that candidate genes can be ranked more easily. Another issue is that the outputs from the Leigh Map are affected by the data inputs, ie the breadth of literature available for individual genes. For example, well-established or common gene defects such as SURF1, POLG, and MT-TL1 are the subject of numerous case reports/publications while newer genes or genes with unclear functions have considerably fewer reports and associated phenotypes.
While there are no current curative therapies for mitochondrial disease, there are numerous compounds which are aimed at symptomatic management, including anti-convulsant drugs and cofactor and vitamin supplements, such as coenzyme Q10, thiamine, and biotin, used to treat corresponding deficiencies.3 The addition of drug targets (a current feature of the MINERVA platform) to the Leigh Map could potentially provide insight into the effectiveness of various agents in treating mitochondrial disease in specific genetic contexts. This is useful for clinical management as certain mitochondrial diseases are potentially treatable.17
The computational nature of the Leigh Map allows for the addition of novel disease genes or phenotypes with relative ease, therefore clinicians have access to a database of all current causative genes, which can enhance the interpretation of WES data. This is especially beneficial within the context of mitochondrial diseases since novel genes are constantly being identified. In Leigh syndrome alone, one third of the causative genes were identified in the last five years.18 Ideally, we will update both the phenotypic and genetic components of the Leigh Map concurrently with the literature. We are also exploring crowdsourcing options from clinicians and machine learning methods to keep the Leigh Map as up-to-date as possible.
Within the domain of mitochondrial disorders there are several phenotypes which have considerable genetic heterogeneity which would also benefit from the creation of specific diagnostic resources, as we have done with the Leigh Map. Our ongoing efforts are to create additional resources for these phenotype groups, including mitochondrial epilepsy, cardiomyopathy, and liver disease. Ultimately, we also aim to create a large comprehensive diagnostic network to encompass the entirety of mitochondrial disease.
The utility of phenomics in IMD diagnosis
Benefits of phenomic databases
The adoption of phenomics into biomedical knowledge bases is beneficial and one can argue essential in the era of NGS. A caveat of WGS or WES outputs are VUS, which can number in the thousands. It is often difficult to disentangle pathogenic variants from variants caused by genetic variation in the population and naturally occurring individual mutations (an average of ∼100 naturally occurring loss-of-function variants per genome).8 The annotation of clinical abnormalities and their integration into the diagnostic pipeline therefore can identify functional groups of genes, which can help to pinpoint truly pathogenic variants and obtain definitive genetic diagnoses.
In recent years several gene-to-phenotype databases and/or knowledge bases have emerged, including large-scale international expert-curated knowledge bases such as ClinVar and ClinGen.19,20 However, there are also several niche expert phenomic resources similar to the Leigh Map, which were developed by experts within a specific disease field to enhance diagnosis and/or clinical management. Such resources have been created for IMDs, Fanconi anaemia, and rare cardiac disorders.21–23 Other tools can be integrated within the bioinformatics pipeline of analysis of WES/WGS which use machine learning and data mining of several clinical and biological knowledge bases to enhance predictive capability.24–27 Resources such as ‘Phenolyzer’ and ‘PHEVOR’, for example, automatically consult several rare disease knowledge bases and, based on text inputs from the user, provide a candidate gene list based on both known gene-phenotype associations and alternative genes within the same gene family.26,27
In the era of digitised medicine, online tools are increasingly being utilised by experts and non-experts alike. Clinicians often use online tools when faced with atypical phenotypic presentations, which is not an infrequent scenario in the context of rare genetic disorders. Patients and their families may also use the internet to access information pertaining to their diagnosis.28 Typical search engines such as Google, while user-friendly, may not be equipped to handle medically specific queries.29 Therefore the increasing presence of high-quality curated user-friendly phenomic tools will help to expedite diagnostic odysseys and also provide education to clinicians, patients and families.
Remaining challenges
The increasing availability of broad and narrow phenomic repositories is an exciting addition to the field of omics. As with all novel technologies, there are a number of emerging challenges. It is important to consider the quality and the number of outputs that phenomic resources provide. Phenomic resources are supplements to and not replacements for clinical and genetic expertise. If a particular disease or genetic defect is associated with a pathognomonic clinical presentation well known to clinicians, it is unlikely that there will be a need to consult a phenomic resource. Conversely, some resources generate hundreds of outputs that may be too large to be manageable or interpreted meaningfully in a short amount of time. A manageable number of outputs is something that is more easily achieved in niche resources such as the Leigh Map, but also needs to be considered in larger resources.
At present, there are few efforts to standardise the collection, annotation, and storage of phenotypic data. This decreases the interoperability between differen t resources and geographical regions. Perhaps in future phenomic resources can benefit from similar standardisation to the American College of Medical Genetics and Genomics variant prioritisation guidelines.30 The annotation of phenotypes in HPO is helpful as there are several thousand phenotypes documented so far. However, there are still several phenotypes, for example biochemical phenotypes, that do not yet have an HPO number and cannot be integrated in resources linked with HPO. This is problematic since these biochemical phenotypes, which are particularly relevant to IMDs, cannot be queried in the aforementioned tools, thus diminishing the utility of these tools for this disease group.31 However, several ongoing projects aim to improve standardisation and interoperability in the phenomics field. One such initiative, harmonising phenomics information for better interoperability in rare disease, aims to create a rare-disease-specific bioinformatics system which integrates expert curated annotations, automated concept recognitions, and standardisation via HPO to maximise the clinically relevant potential of phenomics. Notably the project emphasises the need for standardised data collection and annotation from patient repositories and biobanks worldwide, but these data can be difficult to access and make publically available, especially with the implementation of stringent data protection laws.9,32
There is also the issue of keeping phenomic resources up-to-date. Resources such as the Leigh Map, which are literature dependent, face the challenge of being as current as possible given that novel gene defects are constantly being characterised. Possible solutions that are being explored include expert curation, crowd-sourcing, and automated machine learning methods.9,19,27,29,33⇓
Current live phenomic knowledge bases and tools
Concluding remarks
In recent years, there has been an increase in the utilisation of phenomics to help to diagnose and manage rare diseases, especially those with complex genetic and phenotypic heterogeneity, as observed for many IMDs. The benefits of interactive user-friendly platforms in the interpretation of NGS results are becoming increasingly apparent. While several challenges still need to be mitigated, the advent of phenomics in the IMD field will certainly increase the number of genetic diagnoses, facilitating timely and appropriate clinical intervention, and potentially improving the overall prognosis for affected patients.
- © Royal College of Physicians 2019. All rights reserved.
References








{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.