
ABSTRACT
Henoch–Schönlein purpura (HSP), also known as IgA vasculitis, is a systemic vasculitis which is the most common vasculitis in children. The incidence in adults varies from 3.4 to 14.3 cases per million.1 The classic triad of symptoms include purpuric rash, arthritis and abdominal pain. We present the case of a 20-year-old male with HSP who presented with recurrent episodes of abdominal pain, followed by classical symptoms with an identified post-infectious aetiology.
Case presentation
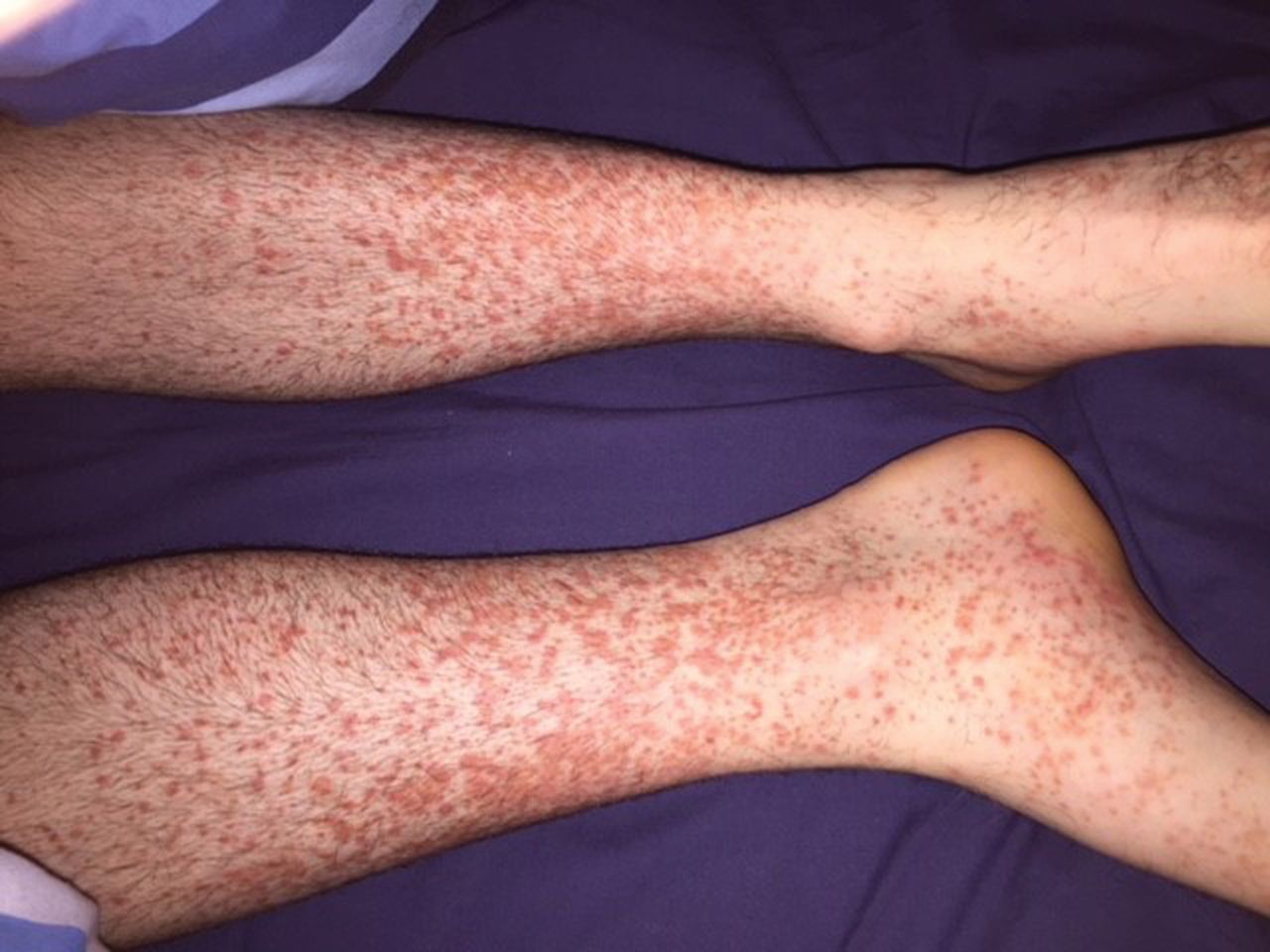
A 20-year-old man with no previous medical history presented with a florid purpuric rash affecting distal extremities, abdominal pain and a monoarthritis of his right wrist. Two weeks prior he had attended the emergency department with a flu-like illness and a swollen tender right ankle, but subsequently self-discharged. Over the course of the following week he began to develop a purpuric rash on his distal extremities (Fig 1), spreading to his buttocks (Fig 2). On presentation he had begun to experience central crampy abdominal pain, increasing in severity over the previous 2 days with no relief from simple analgesia. On initial examination his right wrist was swollen and tender on palpation. He remained apyrexic with a temperature of 36.6°C, and had a blood pressure of 122/66 mmHg, heart rate of 83 bpm and oxygen saturation of 96% on room air. No other obvious infective sources were identified on initial examination with no evidence of meningism. Inflammatory markers were raised with a white cell count of 11.4 × 109/L and C-reactive protein of 36.7 mg/L. Coagulation screen was normal with platelet count 439 × 109/L. Erythrocyte sedimentation rate was elevated at 31 mm/h. Urinalysis revealed a trace of protein.

Purpuric papules of the lower extremities characteristic of Henoch–Schönlein purpura.

Typical rash distribution of Henoch–Schönlein purpura.
Diagnosis and initial management
A working diagnosis of Henoch–Schönlein purpura (HSP) was made and he was commenced on oral prednisolone along with protein pump inhibitor (PPI) cover. Further investigations sent included immunoglobulin (Ig) profile, complement level and albumin creatinine ratio. Antistreptolysin O titre was sent as the patient stated he had sore throat two weeks previous. On day two into admission the purpuric rash was less pronounced, and serum Ig levels revealed an elevated IgA of 5.40 g/L (0.8–2.8 g/L). Over the next 24 hours he began to experience abdominal pain and vomiting with increasing severity requiring opiate analgesia. On examination there was an improvement in purpuric rash, and the abdomen was soft with no guarding or rebound tenderness and bowel sounds were heard. Arterial blood gas revealed a metabolic alkalosis with pH 7.53, bicarbonate (HCO3) standard 29.7 mmol/L and lactate 2.2 mmol/L. Abdominal film showed non-specific gas pattern with no obvious perforation or obstruction. Full blood picture showed a rise in white cell count to 21.4 ×109/L with a prominent neutrophilia. This elevation was felt to be a combination of perfuse vomiting and recent steroid commencement. Given the risk of gastrointestinal (GI) haemorrhage and bowel wall oedema associated with the condition, treatment was changed to course of intravenous (IV) methylprednisolone with IV PPI cover.
Progress
The patient’s abdominal pain and vomiting subsequently improved with a combination treatment of 3 days IV methylprednisolone, IV fluids and antiemetics. Five days after initial presentation, purpuric rash had resolved along with right wrist monoarthritis. Prior to discharge antistreptolysin O titre came back elevated at 400 IU/L and a diagnosis of HSP precipitated by streptococci infection was made. He was subsequently discharged on a slow reducing dose of oral prednisolone, and reviewed in clinic 8 weeks post-admission with a complete resolution of symptoms.
Discussion
HSP is a small vessel vasculitis in which immune complexes of IgA and complement component 3 (c3) are deposited on capillaries, venules and arterioles. In adults, HSP has been observed to have a higher frequency of systemic involvement and the outcome in adults was found to be similar to children with complete recovery from the disease in the majority of cases.2 The typical symptoms include purpuric rash appearing on legs and buttocks however the rash may also be seen on the arms, face and trunk. Joint arthritis involves ankles, knees and elbows and is non-erosive hence does not cause permanent deformity. Forty percent of patients will have kidney involvement, mainly in the form of haematuria which is predominantly microscopic in nature. Proteinuria occurs in more than half of patients involved, some of which is severe enough to cause nephrotic syndrome. Approximately 1% of all HSP patients go on to develop chronic kidney disease, and adults are more likely to do so than children.3
Clinically, since there are no disease-specific laboratory abnormalities, HSP is currently diagnosed based on symptoms, signs and histopathological findings. To date, several sets of diagnostic criteria have been proposed with the most recent gold standard EULAR/PRINTO/PRES criteria 20104 (Table 1).
Diagnostic criteria for Henoch–Schönlein purpura, as developed by EULAR/PRINTO/PRES4
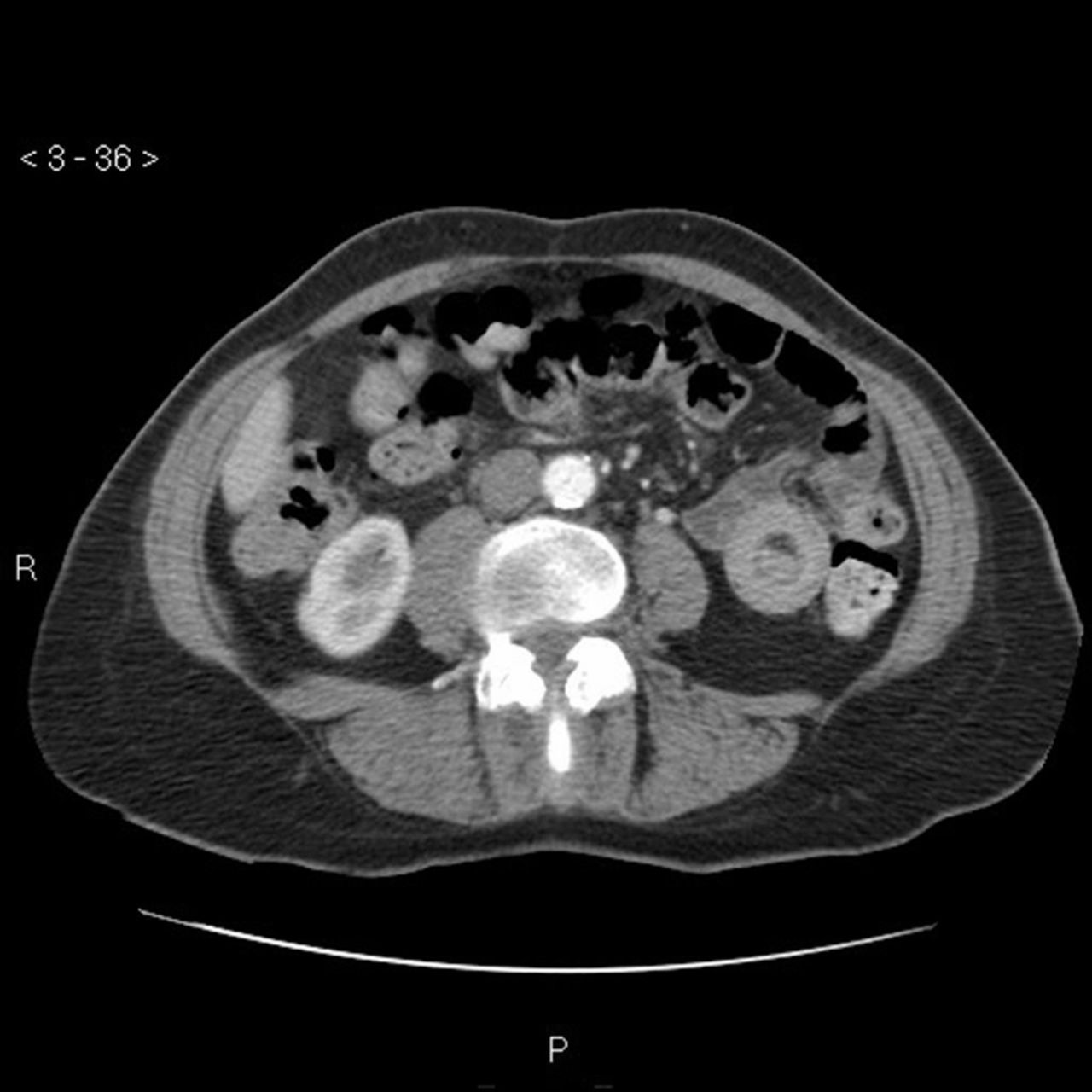
GI symptoms occur in about one half of HSP patients and range from mild symptoms to more significant conditions such as intussusception (Fig 3), GI haemorrhage, bowel ischaemia and perforation. In a study involving HSP in the adult population, 24.1% of patients develop GI symptoms prior to the cutaneous rash.5 Abdominal pain is typically colicky in nature, poorly localized, and can be accompanied with vomiting and bloody diarrhoea.
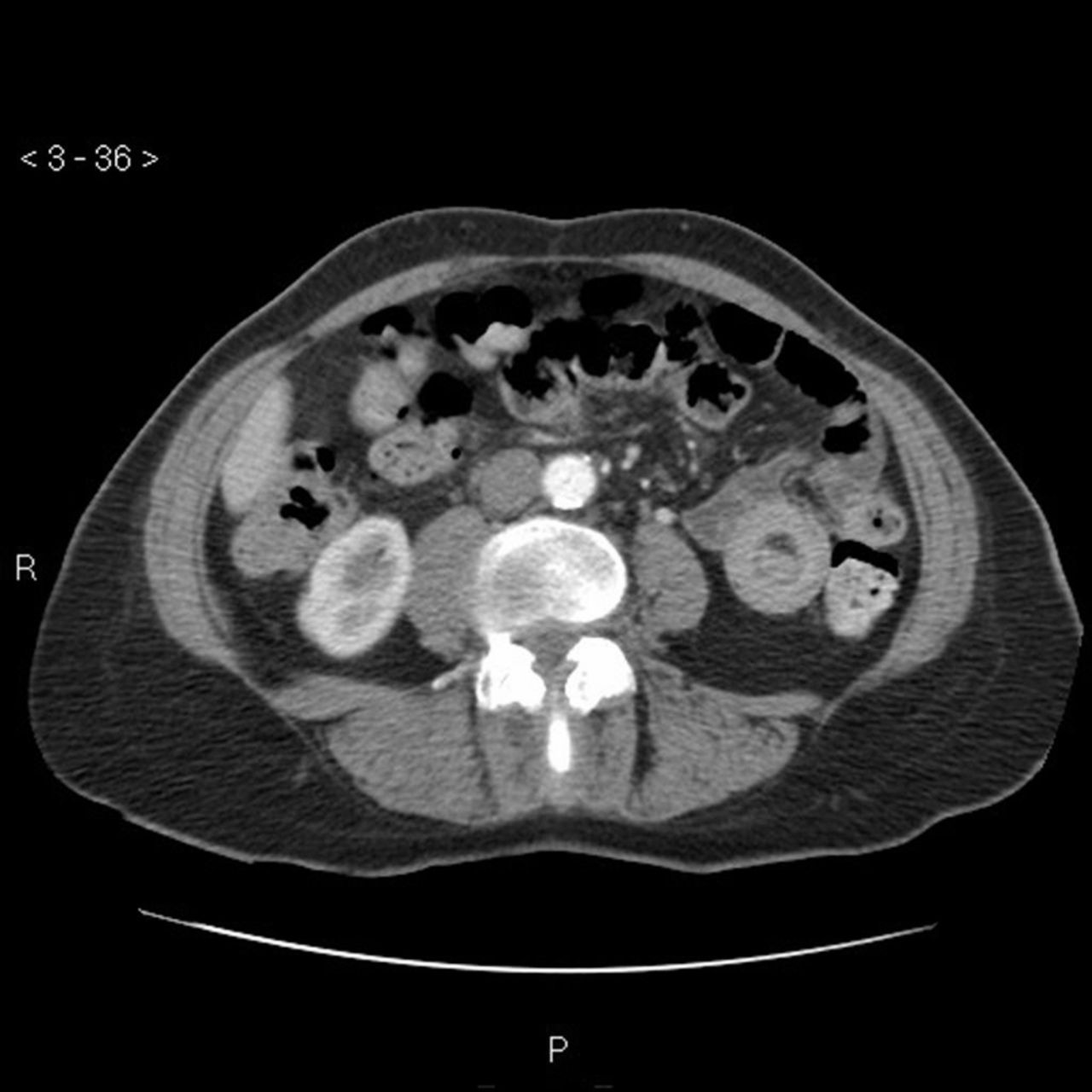
Computed tomography scan of abdomen to show the target sign of intestinal intussusception, also known as the doughnut sign or bull’s eye sign (case courtesy of Dr Sajoscha Sorrentino, Radiopaedia.org, rID: 16879).
Treatment remains primarily supportive in most cases and hospitalisation should be considered for patients with severe abdominal pain, significant GI bleeding or marked renal insufficiency. Supportive measures include adequate hydration and monitoring for abdominal and renal sequelae. Any unnecessary drugs should be discontinued if a drug-related aetiology is being considered. Drugs linked with development of HSP include antibiotics such as vancomycin, cefuroxime, angiotensin converting enzyme inhibitors and anti-inflammatory agents such as diclofenac.5 More than 75% of patients report antecedent upper respiratory tract or pharyngeal infection.6 The development of HSP has been associated with multiple bacterial and viral infectious agents. Examples include group A streptococcal infection, mycoplasma, hepatitis B and herpes simplex virus.
Due to the high spontaneous recovery rate most patients do not receive specific therapy and steroids are generally avoided. Steroids may shorten the duration of abdominal and joint pain but must be weighed up against potential adverse effects. Abdominal symptoms in HSP are caused by haemorrhage and oedema within the bowel wall and mesentery. The pathophysiology of steroid use for GI may be attributed to the reduction in this oedema.
Learning points
HSP is rare in the adult population, and is more commonly found in children
The pathophysiology involves immune complex deposition in organ systems such as skin, joints, kidneys and GI tract
Treatment is mostly supportive however hospitalisation and steroid use should be considered for patients with severe abdominal pain or severe renal insufficiency
Increasing abdominal pain severity should prompt early surgical review for possible bowel ischaemia, perforation or rarely intussusception.
Consent
Patient consent was obtained to write and publish this article.
- © Royal College of Physicians 2019. All rights reserved.








{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.