
ABSTRACT
Pyoderma gangrenosum (PG) is a reactive non-infectious inflammatory dermatosis falling under the spectrum of the neutrophilic dermatoses. There are several subtypes, with ‘classical PG’ as the most common form in approximately 85% cases. This presents as an extremely painful erythematous lesion which rapidly progresses to a blistered or necrotic ulcer. There is often a ragged undermined edge with a violaceous/erythematous border. The lower legs are most frequently affected although PG can present at any body site. Other subtypes include bullous, vegetative, pustular, peristomal and superficial granulomatous variants. The differential diagnosis includes all other causes of cutaneous ulceration as there are no definitive laboratory or histopathological criteria for PG. Underlying systemic conditions are found in up to 50% of cases and thus clinicians should investigate thoroughly for such conditions once a diagnosis of PG has been made. Treatment of PG remains largely anecdotal, with no national or international guidelines, and is selected according to severity and rate of progression. Despite being a well-recognised condition, there is often a failure to make an early diagnosis of PG. This diagnosis should be actively considered when assessing ulcers, as prompt treatment may avoid the complications of prolonged systemic therapy, delayed wound healing and scarring.
Pyoderma gangrenosum
Pyoderma gangrenosum (PG) is a reactive non-infectious inflammatory dermatosis falling under the spectrum of the neutrophilic dermatoses, which includes Sweet’s syndrome and Behcet’s syndrome. The incidence is thought to be approximately 0.63 per 100,000 with the median age at presentation of 59 years.1 The sex incidence ranges from being equal,2 to females being predominantly affected in up to 76% of cases.3 There appears to be a genetic component with cases of PG clustering in families and siblings.4
Clinical features
Classical PG presents most commonly as an extremely painful erythematous lesion which rapidly progresses to a blistered or necrotic ulcer. There is often a ragged undermined edge with a violaceous/erythematous border.1 The lower legs are most frequently affected although PG can present at any body site. The lesion may be precipitated by minor trauma, a phenomenon known as ‘pathergy’.6 Indeed PG lesions are all too often misdiagnosed as simple non-healing ulcers and patients undergo debridement which can result in catastrophic deterioration of the condition through this pathergic response. The condition predominantly affects adults but childhood cases are rarely reported.7 Although there may be some families with PG and inherited syndromes in which PG is a feature, the majority of patients do not have a family history of the condition.
Most cases of PG are of the classic ulcerative type (approximately 85%), but other subtypes include bullous, vegetative, pustular, peristomal and superficial granulomatous variants, with subtypes of PG sometimes transitioning from one form to another.9 The differential diagnosis has to include all other causes of cutaneous ulceration as there are no definitive laboratory or histopathological criteria for PG. PG may also involve extracutaneous sites affecting the eyes (scleritis, corneal ulceration,) lungs (aseptic pulmonary nodules) and the spleen.
Classical PG
The most common form of PG presents as a rapidly progressive painful ulcer with a violaceous undermined edge (Fig 1).
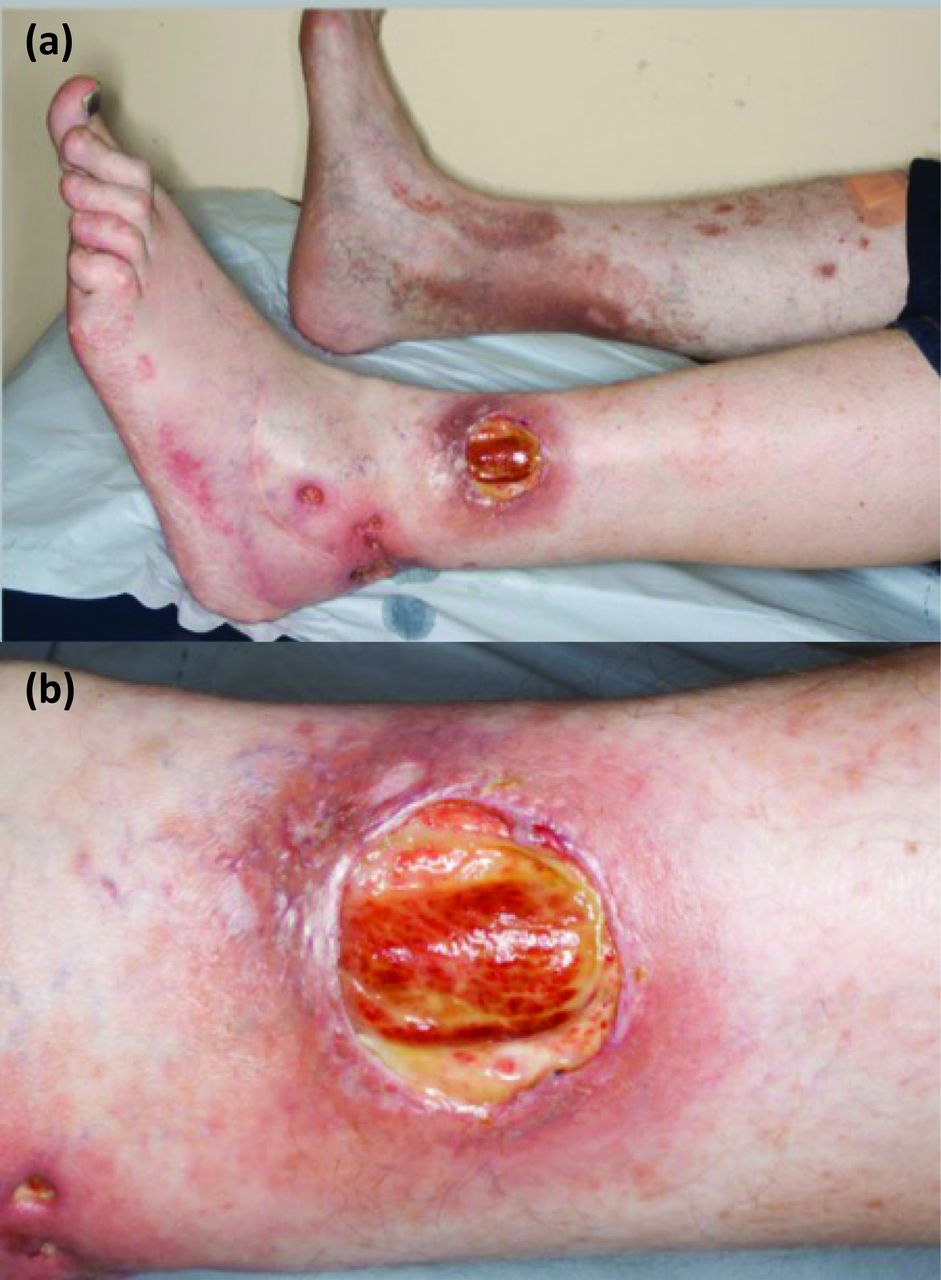
Rapidly progressive painful ulcer
Bullous PG
This form presents with rapidly evolving painful superficial vesicles and bullae arising in waves, often coalescing together most commonly on the arms. Histologically this shares similarities with Sweet’s syndrome. A haematological malignancy should be sought as these are identified in up to 70% cases.10
Pustular PG
This form is most commonly seen in the context of flaring inflammatory bowel disease and presents with painful pustules on a background of erythema, often on the extensor surfaces.11
Granulomatous superficial PG
Otherwise known as vegetative PG, this subtype usually progresses more slowly and presents with verrucous and ulcerative lesions. These patients are less likely to have an underlying systemic condition and do not usually require systemic treatment.12
Peristomal PG
This variant probably results from a pathergic response to trauma from faecal irritation or secondary to appliances on the skin and is most frequently seen in the context of stomas in patients with inflammatory bowel disease.13
Malignant pyoderma
This is an important but rare clinical variant that presents with destructive ulceration typically affecting the upper torso, head and neck. Lesions do not display the violaceous edge seen in classical PG and the condition is not associated with systemic disease.15
Aetiology
Underlying systemic conditions are found in up to 50% of cases and so it is imperative to try and identify such conditions once a diagnosis of PG has been made.14 Most frequently associated conditions include inflammatory bowel disease in up to 30% of cases, rheumatoid arthritis and seronegative arthritides in up to 10% of cases, haematological malignancies or monoclonal gammopathies (in particular immunoglobulin A gammopathy) in 5% of cases and other malignancies in 5%. Other systemic conditions have also more rarely been associated with PG, including those of chronic infection and inflammation.1,16 Drugs such as propylthiouracil, tyrosine kinase inhibitors, TNFα inhibitors and granulocyte-colony stimulating factor have been implicated but the underlying disease for which the medication has been prescribed may be the triggering factor.17
PG associated syndromes have been described and these include PG with cystic acne and hidradenitis suppurativa (PASH),18 PG with pyogenic arthritis and acne (PAPA),19 and PG with pyogenic arthritis, acne and hidradenitis suppurativa (PAPASH).20
Pathophysiology
The pathogenesis of PG remains unclear however it is recognised that neutrophils play a key role in the disease process.18 Upregulation of a number of key proinflammatory and neutrophil chemotactic factors within lesional skin have been identified and these include IL-1β, IL-17, TNFα, IL-8, IL-6, IL-17 and IL-23. IL-8 has been demonstrated to produce PG in animal models,22 it is also induced in fibroblasts of PG ulcers23 and its associated ligands are over-expressed in PG.24 There is also increased matrix metalloproteinase (MMP) expression, in particular MMP 9 and 10 which could contribute to poor healing25 together with clonal expansion of T-cells being identified in the skin and serum of patients with PG.26 However, the exact role of lymphocytes in the pathogenesis of PG has yet to be elucidated.
A genetic basis for PG has so far been documented in the syndromic presentations with mutations in the PSTP1P1/CD2BP1 gene in PAPA and PASH syndromes. Normally pyrin inhibits inflammasome activation but the PSTPIP1 mutant inhibits the anti-inflammatory effect of pyrin and this results in the release of pro-inflammatory cytokines.27 The results of more sophisticated exome sequencing and next generation sequencing in other patients with PG are awaited.
Histological Findings
PG remains a clinical and sometimes challenging diagnosis and although histology of skin biopsies can be supportive, the main value of the skin biopsy is to exclude other causes of cutaneous ulceration and to allow specimens to be sent for bacterial, mycobacterial and fungal cultures. The biopsy should include the active border of the ulcer and penetrate deep to subcutaneous tissues. Patients should be warned that such a surgical procedure will invariably cause an enlargement of the ulcer, as well as potentially inducing a pathergic immunological response to trauma.
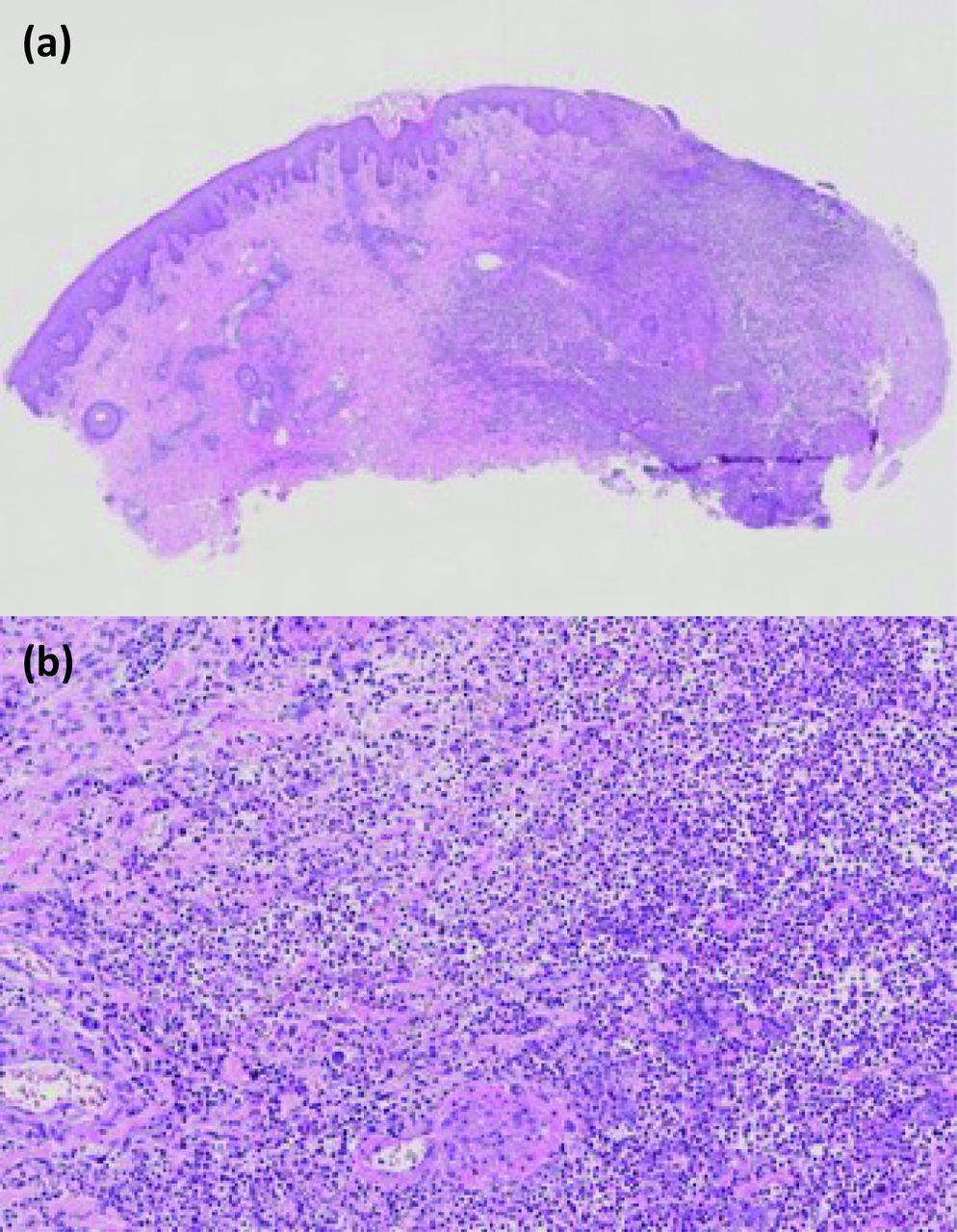
The histological findings can be variable and depend on the site of the biopsy and age of the lesion. In classic ulcerative PG there may be ulceration of the epidermis and dermis associated with an intense neutrophilic infiltrate, neutrophilic pustules and abscess formation (Fig 2).28 Different histological findings will be seen depending on the clinical variant. Vasculitis is sometimes identified histologically but this may be secondary to the ulceration. If identified, causes of true vasculitis and infection should be investigated.29

Histological findings with an intense neutrophilic infiltrate, neutrophilic pustules and abscess formation
Diagnosis
Thus far there are no validated, established diagnostic clinical or pathological criteria to diagnose PG. Su et al have proposed a diagnostic tool requiring two major and two minor criteria (see Table 1), maintaining PG as a diagnosis of exclusion.30 More recently, Maverakis et al have proposed new criteria based on a consensus of international experts, requiring one major and four minor criteria (see Table 2).31 This is yet to be widely adopted, but no longer renders PG as a diagnosis of exclusion which may thus provide an improved diagnostic tool.
Diagnostic tool for pyoderma gangrenosum
Improved diagnostic tool for pyoderma gangrenosum
Differential diagnosis
Other causes of cutaneous ulcers should be considered. These include arterial and venous disease, haematological causes (sickle cell disease, cryoglobulinaemia, anti-phospholipid syndrome), vascular occlusion, vasculitis, infections, calciphylaxis, drug-induced ulceration, primary or metastatic tumours, hypertension (Martorell ulcer) and other inflammatory disorders including cutaneous Crohn’s disease.
How to approach the patient
A thorough history is key with specific enquiry regarding possible pathergic response to minor or major trauma, as well as a history of pain, rapid progression, symptoms suggestive of infection or systemic disease and a detailed drug history. Clinical examination of the ulcer as well as a full body examination is essential and a skin biopsy as suggested above.
Laboratory investigations should include a full blood count, erythrocyte sediment rate, C-reactive protein, liver and renal function tests, protein electrophoresis, urinary Bence Jones protein, a full hepatitis screen, a vasculitic screen, cryoglobulins if the history is suggestive and a coagulation screen to investigate for thrombotic causes of ulceration. A faecal calprotectin is advised if there is a clinical suspicion of inflammatory bowel disease. A chest x-ray is also indicated as a baseline screen prior to systemic therapy and computed tomography (CT) if there is a suspicion of underlying malignancy.
Treatment
Treatment of PG remains largely anecdotal with only two randomised controlled trials (RCT), and is therefore based largely on case series and poorly evidenced publications.32,33 With no national or international guidelines, management is therefore challenging, and treatment choice is based on the severity and extent of the PG (Fig 3).34
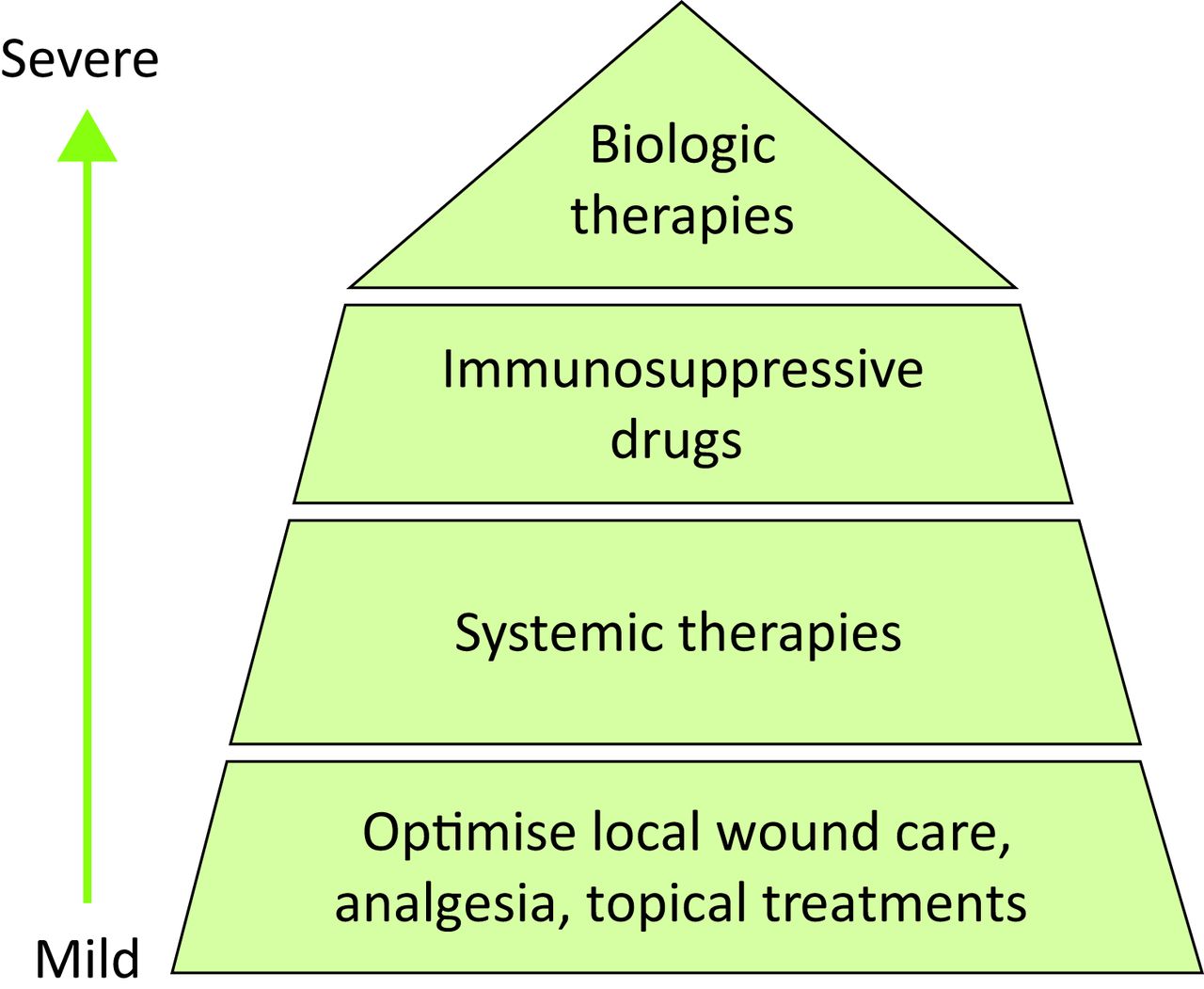
Treatment choice based on the severity and extent of pyoderma gangrenosum
First-line treatment is aimed at optimising local wound care, particularly important in cases of PG arising on the leg, where wound healing can be delayed by vascular disease. Supportive therapy with appropriate dressings, compression (if arterial insufficiency has been excluded) and adequate analgesia are all essential to optimise healing. Potent topical corticosteroids and tacrolimus ointment applied to the ulcer surface are useful and intralesional injections of corticosteroid into the erythematous active border may be considered.35
In more severe disease, systemic therapy is required. Oral corticosteroids (0.5–1 mg/kg/day) are the mainstay of treatment and are used to gain rapid control. Ciclosporin can be used either alone or in combination with corticosteroids as a steroid-sparing agent, in cases where prolonged treatment is required.36 In a multicentre RCT of 121 patients, prednisolone 0.75 mg/kg/day (maximum dose 75 mg) was compared to ciclosporin 4 mg/kg/day (maximum dose 400 mg) and only 50% of patients achieved remission at 6 months and there was no significant difference between the two monotherapies.32 Other systemic treatments utilised with varying success include colchicine, sulphasalazine, dapsone, minocycline, apremilast and thalidomide. Pulsed intravenous methylprednisolone can be useful in initiating a rapid response given together with immunosuppressive drugs such as methotrexate, mycophenolate mofetil, cyclophosphamide, azathioprine and high dose intravenous immunoglobulin.
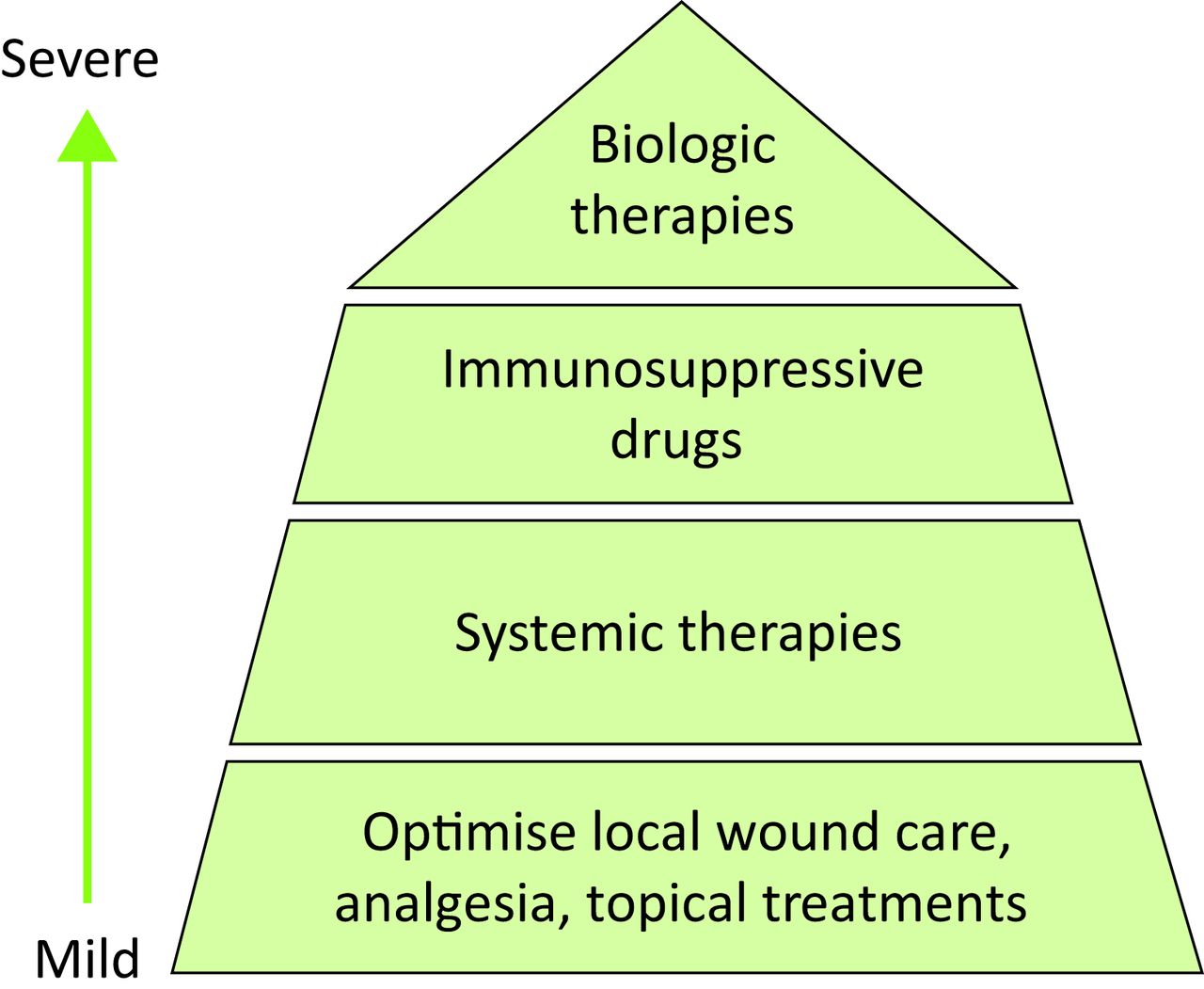
There is now a growing body of evidence to support biologic therapy as a treatment of PG37 targeting a number of cytokines but there is no consensus as to whether these treatments should be administered only when other treatments have failed or as first line therapies in severe PG (Fig 4).

Biologic therapy as a treatment of pyoderma gangrenosum targeting a number of cytokines
Thus far infliximab has the largest body of evidence to support its early use in PG. In a RCT investigating the use of infliximab (5 mg/kg intravenous) versus placebo, 69% of patients demonstrated clinical improvement at week six after just one infusion.38 A retrospective observational study has shown faster wound healing in a significant proportion of patients treated with infliximab and adalimumab versus oral corticosteroids alone,39 further supporting the argument that biologic therapy should be considered as an early treatment strategy. Adalimumab has shown efficacy in achieving wound healing in recalcitrant cases40 and there are case reports demonstrating success with etanercept,41 ustekinumab,43 anakinra43 and canakinumab.44
Summary
Despite being a well-recognised condition there is often a failure to make an early diagnosis of PG. It is important for all clinicians to be aware of this condition and to actively consider PG when assessing patients with ulcers, as appropriate and prompt treatment at an early stage of the disease may avoid the complications of prolonged systemic treatment, delayed wound healing and scarring.
- © Royal College of Physicians 2019. All rights reserved.








{kind=link}
{kind=link}
{kind=link}
{kind=link}