
ABSTRACT
We present the case of an 83-year-old woman, with known asthma, admitted with increasing dyspnoea, wheeze and a productive cough. In addition to maintenance inhaled therapy, the patient was also on long-term mirtazapine and furosemide. Following acute treatment with nebulised salbutamol she became increasingly dyspnoeic and developed a metabolic acidosis with a significantly raised blood lactate level. After cessation of ß2-adrenergic medication, the patient's clinical condition improved with resolution of her lactic acidosis; salbutamol induced lactic acidosis was diagnosed. This clinical scenario is common but not well described. Here we discuss the mechanisms, investigation and management of raised serum lactate and lactic acidosis in the context of acute asthma and the possible interactions of polypharmacy and comorbidities in the acute medical setting.
Key learning points
Knowledge of lactate metabolism is needed to diagnose potential causes of lactic acidosis.
ß2-agonist therapy should be considered in the differential diagnosis of lactic acidosis.
Comorbidities and polypharmacy may increase risk of salbutamol induced lactic acidosis.
Over-investigation and treatment of salbutamol induced lactic acidosis may potentially cause patient harm.
Case presentation
An 83-year-old woman, with longstanding asthma, was admitted via the emergency department with a 2-day history of shortness of breath, generalised wheeze and productive cough with green sputum. Her maintenance asthma therapy was inhaled symbicort 400/12 twice daily, formoterol fumarate 12 μg twice daily, and terbutaline sulphate 500 μg as required. She had no admissions with asthma in the previous 12 months and had never required intensive care admission. She had never smoked. Other medications were a cyclic antidepressant and a loop diuretic. Initial observations: heart rate was 115 beats per minute (bpm), respiratory rate was 27 breaths per minute, oxygen saturation (SpO2) was 93% on air and peak flow was 160 L/min (predicted 320 L/min). Auscultation revealed diffuse bilateral wheeze. Oxygen was commenced via 35% Venturi mask (FiO2 0.35). Chest X-ray demonstrated hyperexpanded lung fields but no focal pathology. Arterial blood gas (ABG) result on FiO2 0.35 is shown in Table 1. At this stage, lactate was 1.4 mmol/L (0.5–1.6). Blood results were white cell count of 10.7 × 109/L (4–11), neutrophil count of 8.6 × 109/L (2–7.5), eosinophil count of 0.55 × 109/L (0.00–0.40), eGFR of >90 mL/min/1.73m2 (>90) and C-reactive protein of 13 mg/L (<5).
Arterial blood gas trends during acute admission
Acute severe asthma presumed secondary to lower respiratory tract infection was diagnosed and the patient initially treated with intravenous magnesium sulphate (2 g), nebulised salbutamol (5 mg), oral prednisolone (30 mg) and oral doxycycline (200 mg) stat in addition to controlled oxygen. One hour later, upon review by the respiratory team, intravenous aminophylline loading dose followed by infusion was commenced and the frequency of salbutamol nebulisers was increased.
Two hours post admission despite ‘back-to-back’ nebulised bronchodilator therapy, the patient's observations continued to deteriorate. Oxygen saturations remained >96% on 35% oxygen but the patient's dyspnoea had subjectively worsened. She was now tachypnoeic (>36 breaths per minute), tachycardic (>140 bpm) and hypertensive (systolic BP >200 mmHg). At this stage, the intensive care team reviewed the patient. Repeat ABG (FiO2 0.35) demonstrated improvement of oxygenation but worsening base deficit and significant elevation in lactate to 6.8 mmol/L (Table 1). Clinically the patient had reduced wheeze on auscultation and adequate oxygenation, suggesting life-threatening asthma was unlikely to be the cause. On further direct questioning the patient stated that she had lower abdominal pain which was chronic yet not previously investigated. The differential diagnosis for the lactic acidosis included bowel ischaemia or secondary to ß-adrenergic stimulation from salbutamol therapy. Salbutamol nebulisers were discontinued, computed tomography (CT) of the abdomen and pelvis (without contrast) was requested and surgical opinion sought.
Over the next 4 hours, the patient's observations progressively improved. The CT revealed moderate uncomplicated sigmoid diverticular disease but no other pathology. Repeat blood gasses (5 hours post admission, FiO2 0.28) demonstrated resolution of acidosis with a drop in lactate to 2.0 mmol/L (Table 1). A diagnosis of salbutamol induced lactic acidosis (SILA) was made and further investigations deemed unnecessary. The lactate returned to normal range over the next 2 days. The patient was discharged on day 3 with early outpatient follow-up in the asthma clinic.
Discussion
SILA is recognised anecdotally in clinical practice but is rarely formally diagnosed. In acute medical admissions raised lactate levels without acidosis (lactataemia) and lactic acidosis are common clinical scenarios. These patients frequently have advanced age, multiple comorbidities, and may be prescribed medications which increase the risk of lactataemia and lactic acidosis (Table 2).1–3 The acute prescription of ß2-adrenergic agonists is also common and another independent risk factor for lactataemia.3,4 Thus an awareness of underlying mechanisms of lactataemia is important for physicians in order to identify, monitor and appropriately treat these patients.
Mechanisms and causes of lactic acidosis
Lactic acidosis is often classified into types A and B based upon the presence, or absence, of tissue hypoxia but may occur due to both hypoxic and non-hypoxic factors concurrently.3,5 Five common mechanistic groups have been proposed (Fig 1; Table 2).3
Glycolysis produces pyruvate which, under aerobic conditions, is metabolised by the tricarboxylic acid (Kreb's) cycle and oxidative phosphorylation. Increased glycolysis produces increased amounts of pyruvate, which is metabolised to lactate anaerobically when aerobic pathways are overwhelmed.
The pyruvate dehydrogenase complex links glycolysis to the tricarboxylic acid cycle and oxidative phosphorylation; inhibition causes pyruvate and thus lactate accumulation.
Insufficient oxygen supply to meet tissue demand promotes anaerobic respiration.
Impairment of oxidative phosphorylation.
Impairment of lactate metabolism and excretion. This occurs predominantly by the liver (~70%), kidneys and other tissues also contribute.
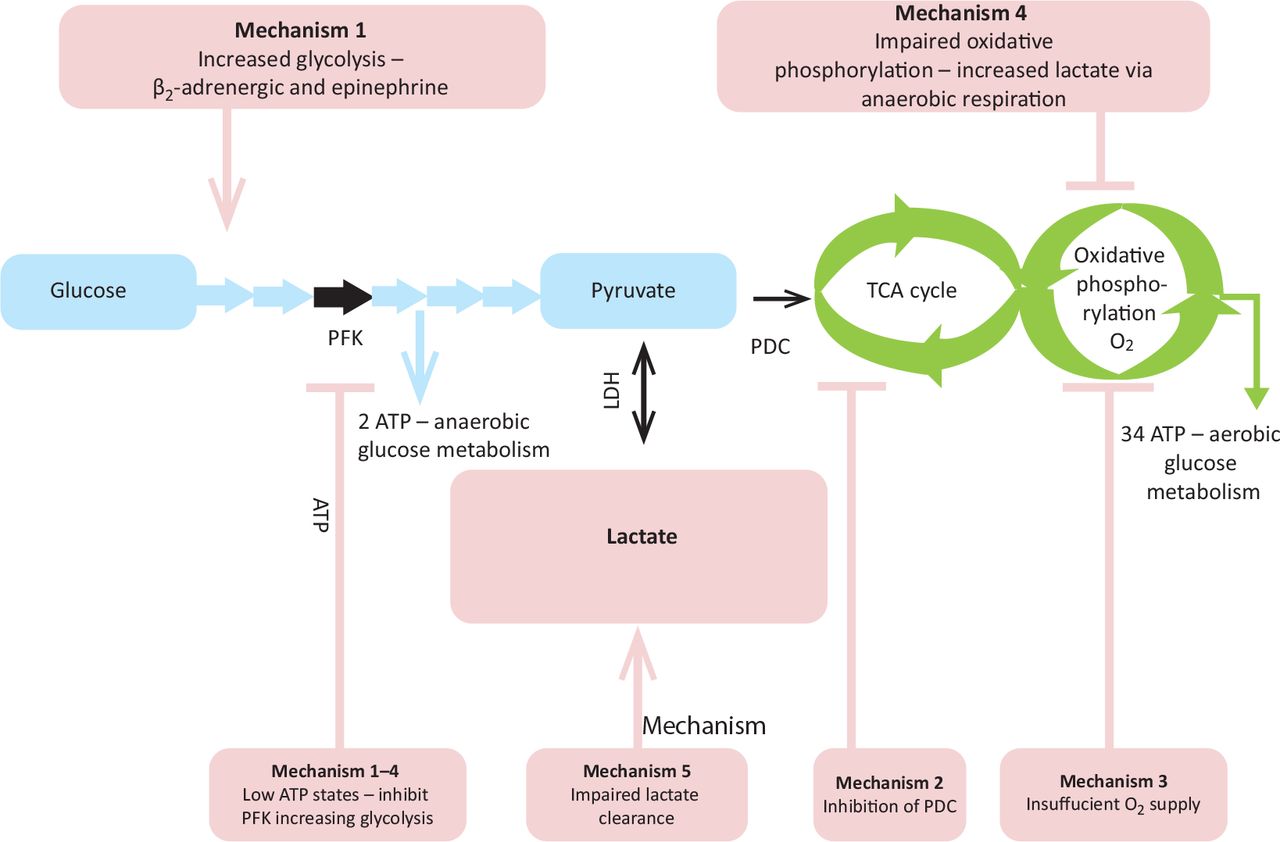
Glycolysis pathway and mechanisms of increased serum lactate. Glycolysis pathway in light blue. Mechanisms of lactate production in light red. Aerobic respiration in green. Black arrows are key enzymatic steps. ATP = adenosine triphosphate; LDH = lactate dehydrogenase; PDC = pyruvate dehydrogenase complex; PFK = phosphofructokinase (rate limiting enzyme in glycolysis); TCA = tricarboxylic acid cycle.
Sporadic case reports of SILA in adults with severe asthma date from 1985.4,6 ß2-agonists deplete adenosine triphosphate levels by enhancing Na+/K+ pump activity and directly increase glycolysis through adrenergic stimulation (mechanism 1).3,5 The resultant high lactate state requires enhanced lactate metabolism and clearance; when this is overwhelmed lactataemia and SILA may develop. SILA usually responds rapidly to cessation of ß2-agonists, with the body's normal pathways excreting excess lactate. Literature suggests a 2–6 hour interval for repeat serum lactate measurements and monitoring until resolution.3
Multiple mechanisms of lactataemia may occur in the same patient.6–9 Although admission serum lactate was normal in this patient, multiple factors including hypoxia, ß2-agonist use, infection and dehydration all required potential consideration and treatment. The patient was prescribed a cyclic antidepressant and loop diuretic; both may increase lactate levels by impairing renal function or oxidative phosphorylation, respectively (Table 2; Fig 2). There is little literature on the cumulative effect of age, comorbidities and medications to SILA risk.3,6–9 A recent review highlighted the array of causative medications and severe morbidity of hyperlactataemia.10 Table 2 and Fig 2 highlight medications which may cause lactataemia and need consideration when initiating salbutamol therapy.

Mechanisms by which selected medications cause hyperlactataemia. Aerobic respiration in green. Red ‘X’ is inhibition. Three red ‘+’ is stimulation. Red bar is circulation. FAD+ = oxidised flavin adenine dinucleotide; FADH2 = flavin adenine dinucleotide hydroquinone; LDH = lactate dehydrogenase; NAD+ = oxidised nicotinamide adenine dinucleotide; NADH = reduced nicotinamide adenine dinucleotide; NRTIs = nucleoside reverse transcriptase inhibitors; TCA = tricarboxylic acid cycle.
In this acute asthmatic, salbutamol was undoubtedly the main cause of lactic acidosis, however it is likely that her maintenance medications and age-related decline in metabolism and excretion were additive factors in the development of lactic acidosis.
Conclusion
An understanding of the mechanisms of lactataemia is required to investigate, diagnose and manage SILA. In patients with multiple comorbidities and polypharmacy, there are many potential causes of lactic acidosis.
Consent
Written consent was not sought. The clinical presentation is non-specific and every effort has been made to remove or mask patient identifiable information and protect patient anonymity.
Acknowledgements
Dr Laurence Pearmain is in receipt of a clinical research training fellowship funded by the Medical Research Council.
- © Royal College of Physicians 2020. All rights reserved.








{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.