
Case presentation
A 45-year-old woman, referred for assessment of a chronic dry cough, underwent plain chest X-ray which demonstrated innumerable small pulmonary nodules producing a ‘sandstorm’ appearance (Fig 1). She was constitutionally well and not dyspnoeic. The cough had subsided considerably by the time of her review. Tests for active tuberculosis, autoimmune disorders and thyroid disease were negative. High-resolution computed tomography showed widespread interlobular septal thickening and a dense micronodular pattern consistent with pulmonary alveolar microlithiasis (PAM; Fig 2).
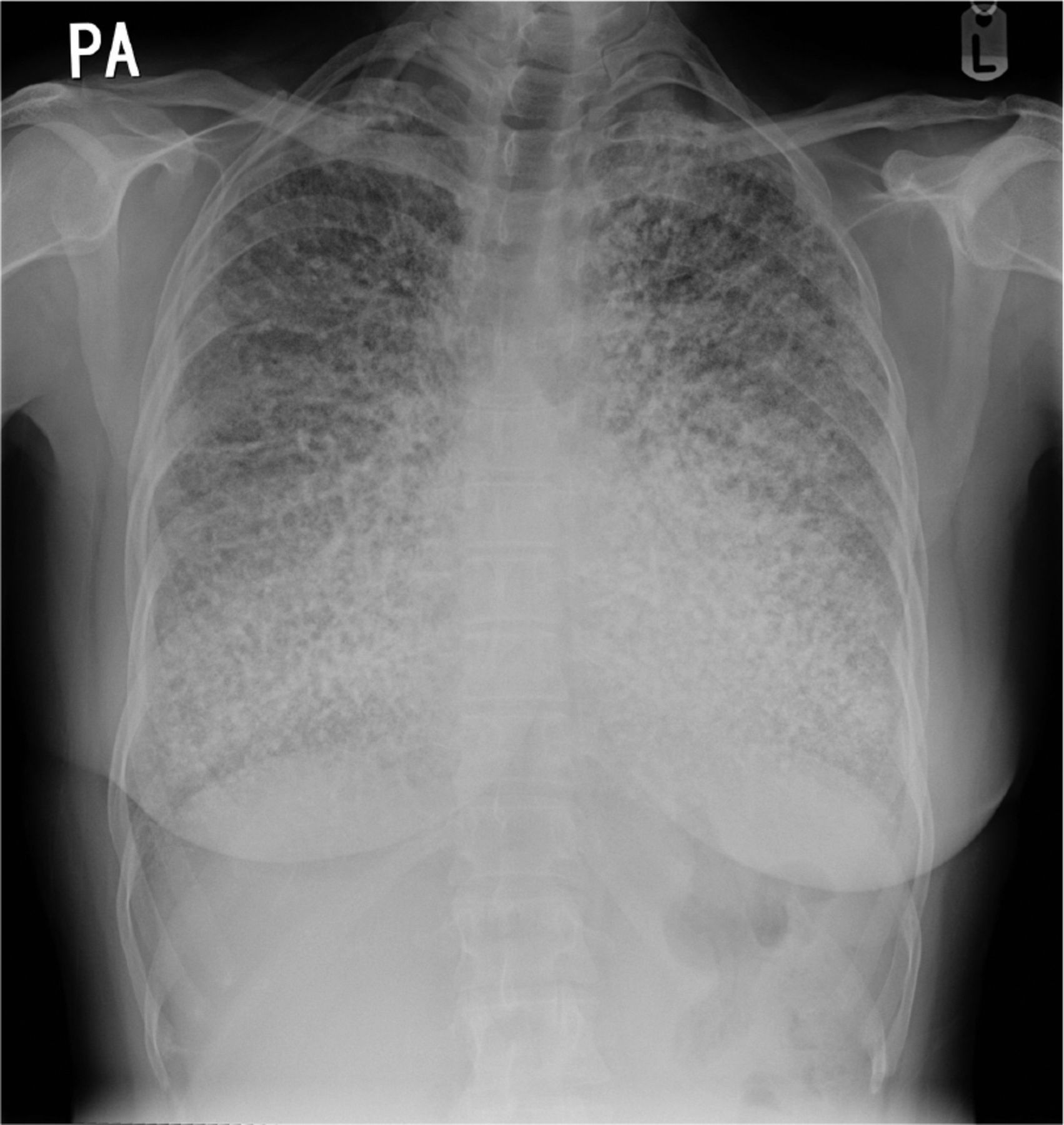
Chest X-ray showing widespread pulmonary calcification suggesting a ‘sandstorm’ appearance.

Axial lung window high-resolution computed tomography. a) Axial lung window setting demonstrating extensive interlobular septal thickening and a micronodular pattern. b) Axial wide-window setting reveals extensive high-density calcified spherules consistent with pulmonary alveolar microlithiasis.
Genetic testing confirmed that the patient was homozygous for a pathogenic frameshift variant in the SLC34A2 gene: c.1653_1660del, which results in a termination codon, p.(Trp552AlafsTer109). The protein encoded by this gene is involved in phosphate homeostasis and alveolar surfactant metabolism. Although a family history of lung disease was not obtained, she reported parental consanguinity.
Pulmonary function testing demonstrated a mild restrictive ventilatory defect with slight gas transfer impairment. Regular monitoring was instituted and a referral made to the regional thoracic transplant assessment service.
Discussion
PAM is a rare autosomal recessive disease characterised by the accumulation of innumerable calcium phosphate deposits known as microliths in the lungs. It is caused by pathogenic variants in the SLC34A2 gene which is located on chromosome 4p15 and encodes a 690-amino acid type IIb sodium phosphate cotransporter. This protein is expressed in alveolar type II cells and its mutation results in disordered phosphate homeostasis and clearance from the alveoli.1
The extensive ‘sandstorm’ calcification in PAM is typically an incidental X-ray finding which precedes the onset of significant symptoms.2 Genetic testing confirms PAM, a diagnosis usually made in childhood to the 4th decade of life. Familial occurrence is seen in a third of cases and there is also a strong association within consanguineous families.1
While patients are often minimally affected at the time of diagnosis, regular surveillance is important as progression of the condition can result in ventilatory failure secondary to extensive microlithiasis, pulmonary fibrosis, pneumothorax or cor pulmonale.2 To date, lung transplant remains the only effective therapeutic option.
- © Royal College of Physicians 2020. All rights reserved.
References








{kind=link}
{kind=link}
Related Articles
Cited By...
- No citing articles found.