
ABSTRACT
In recent years, autoimmunity has been increasingly recognised as an important cause of encephalitis. Many different antibodies are now known to target antigens on the neuronal surface, and some of these are associated with characteristic clinical presentations, although seronegative cases are also recognised. Autoimmune encephalitis may mimic other conditions, including primary psychiatric disorders, particularly early in the disease. Because early immune treatment of autoimmune encephalitis improves patient outcomes, and indeed many make a good recovery, it is important to recognise these syndromes promptly.
Key points
Autoimmune encephalitis should be a key part of the differential diagnosis in patients with alterations in cognition, consciousness, personality or behaviour.
Antibody testing should be guided by recognisable clinical syndromes, but also should account for the expanding numbers of recognised antibodies and overlapping phenotypes.
Autoimmune encephalitis may present sub-acutely with normal or subtly abnormal cerebrospinal fluid findings and neuroimaging.
Despite severe symptoms and long intensive treatment unit stays, the outcome of autoimmune encephalitis is good in most cases if early immune therapy is given.
The optimum first- and second-line treatment strategy for autoimmune encephalitis is unclear and well-designed clinical trials are needed.
Introduction
Encephalitis, a disease of inflammation of the brain, may be caused by an infectious pathogen or by autoimmune processes. Over the last 5–10 years, encephalitis associated with antibodies against neuronal surface antigens has been increasingly recognised.
Autoimmune encephalitis is thought to account for at least 20% of cases of encephalitis, therefore the UK might expect approximately 1,000 cases/year.1,2 Many patients with autoimmune encephalitis (AE) have the capacity to make a good recovery if treated appropriately, despite a stormy disease course. Therefore, it is important to have a high index of suspicion to recognise cases early. However, the myriad antibodies now available for testing have the potential to cause confusion.
We explore recent developments in AE research and practical implications for physicians. Antibodies associated with intracellular antigens are often paraneoplastic and immune-therapy responses may be limited. Also, some antibodies, such as thyroid peroxidase antibodies, are thought to represent an epiphenomenon. Therefore, we will focus on encephalitis in adults with antibodies directed against neuronal surface antibodies, where early diagnosis and management can substantively improve outcomes.
Clinical syndromes
The spectrum of clinical presentations associated with AE is wide and partly reflects the antibody involved (Table 1). The most recognisable clinical syndromes are limbic encephalitis and NMDA-R (N-methyl-D-aspartate receptor) encephalitis.
Key features of syndromes associated with neuronal surface antibodies
Limbic encephalitis, reflecting inflammation of the medial temporal lobe, may present with changes in behaviour, seizures or memory difficulties. Two of the most common and well-characterised antibodies which are associated with limbic encephalitis are those directed against leucine-rich glioma-inactivated 1 (LGI1) and contactin-associated protein-like 2 (CASPR2). AE associated with LGI1 antibodies generally affects older patients and is unusual in those <40 years old. LGI1 antibodies may be associated with faciobrachial dystonic seizures: brief jerking movements affecting ipsilateral arm and face, which are highly specific to LGI1 and may precede the onset of encephalitis, but may not be present in many.3 Hyponatraemia is also a common finding.4 CASPR2 antibody encephalitis can present with a crossover of central and peripheral nervous system features including memory difficulties, sleep disturbances and peripheral nerve hyperexcitability syndromes. Testing for voltage gated potassium channel (VGKC)-complex antibodies should now be abandoned in favour of testing for LGI1 and CASPR2 antibodies, which target proteins associated with the ion channel. VGKC positivity in the absence of LGI1 or CASPR2 antibodies has been reported in a heterogenous group of clinical syndromes, but is not thought to be a true marker of disease.5
NMDA-R encephalitis most often affects children and younger adults, has a 4:1 female predominance and is associated with ovarian teratoma in around half of females. The disorder begins with abnormal behaviour, often with psychotic or affective features, developing by 1 month into a characteristic clinical picture. This may include seizures, movement disorder (classically orofacial dyskinesia, but limb dyskinesia/chorea is often predominant), reduced consciousness level and autonomic dysfunction (particularly bradycardia/tachycardia, hyperthermia and fluctuating blood pressure).6 NMDA-R encephalitis can occasionally be triggered by herpes simplex virus (HSV) encephalitis: in a recent prospective study of 51 patients with HSV encephalitis, 14 developed secondary autoimmune encephalitis associated with neuronal antibodies, although three also developed NMDA-R antibodies in the absence of clinical evidence of encephalitis.7
Other, more recently described syndromes are associated with antibodies against γ-aminobutyric acid receptors (GABAR) type A or B, the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) and dipeptidyl-peptidase-like protein 6 (DPPX; Table 1).
Clinicians should be aware of formes frustes within these classical syndromes, for example, some patients with incipient NMDA-R encephalitis may present with first episode psychosis or prominent movement disorder.
Diagnosis
Patients presenting with symptoms suggesting encephalitis should be admitted urgently and undergo lumbar puncture and neuroimaging. The main concern at the outset is to exclude an infective cause such as HSV, which can cause rapidly progressive brain oedema, necrosis and death. Aciclovir treatment in HSV encephalitis is a life-saving intervention. Several antibody-associated encephalitides, particularly NMDA-R encephalitis, can present similarly to primary psychiatric illness. ‘Red flag’ early features for AE include an infective prodrome, rapid progression, movement disorder, focal neurological signs, seizures or unexplained hyponatraemia.8
Although autoimmune encephalitis is now usually a positive diagnosis, other mimics of AE should be considered, including primary central nervous system (CNS) lymphoma, neurosarcoidosis, CNS vasculitis, tumours, genetic epilepsy syndromes, mitochondrial encephalomyopathies and prion disease.
CSF findings are often abnormal in AE, with a mild lymphocytic pleocytosis and/or moderately raised CSF protein. However, up to a third of patients have a completely normal CSF and so this does not exclude the diagnosis.9
Neuroimaging with magnetic resonance imaging (MRI) is essential, although this may also be normal in up to a third of cases.10 In general, MRI findings are nonspecific, although medial temporal lobe signal change frequently occurs in limbic encephalitis (eg Figs 1a and b).
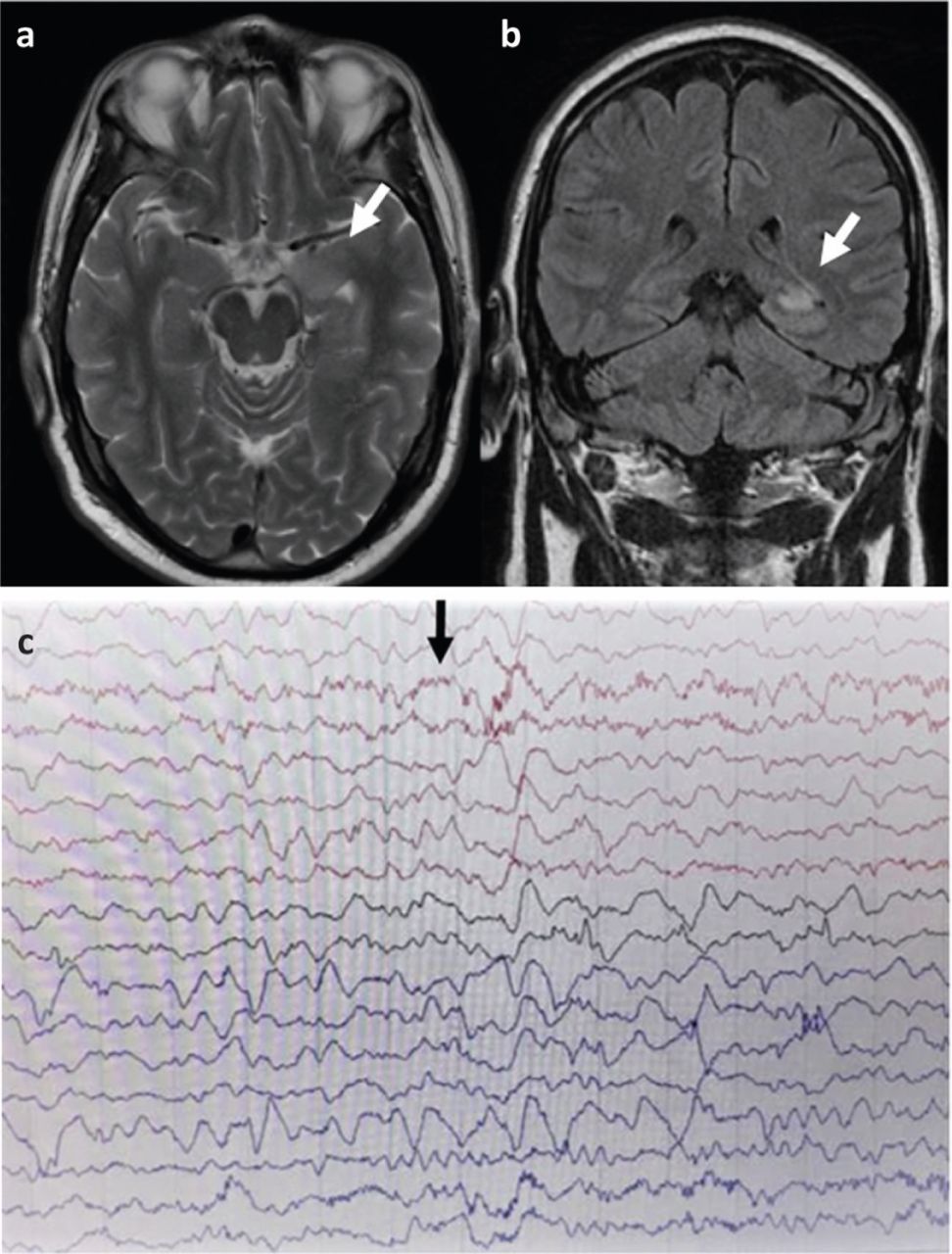
a) T2 weighted axial magnetic resonance imaging LGI-1 antibody encephalitis showing high signal in left medial temporal lobe. b) Coronal T2 fluid-attenuated inversion recovery magnetic resonance imaging showing high signal in the left medial temporal lobe. c) Electroencephalography with N-methyl-D-aspartate receptor antibody encephalitis showing characteristic finding of extreme delta brush.
Electroencephalography (EEG) often identifies encephalopathic changes that can support the diagnosis but are nonspecific. Caution is required as psychiatric and antiepileptic drugs can cause similar features.11 EEG can help to diagnose non-convulsive status epilepticus or to distinguish seizures (eg epilepsia partialis continua (EPC)) from a movement disorder. NMDA-R encephalitis may be associated with the characteristic EEG pattern of extreme delta brush (Fig 1c).
Antibody testing for encephalitis is a rapidly evolving area of neuroimmunology. Certain autoimmune encephalitides, particularly those associated with NMDA-R and LGI1 antibodies, present with recognisable clinical syndromes and diagnostic suspicion may already be high so that antibody testing can be targeted. In other cases that do not have distinguishing features, testing for a wider range of antibodies may be necessary, in consultation with a neurologist and neuroimmunology laboratory. Panels of immunofluorescence tests have been developed commercially and their role is still evolving, considering the balance between increased chance of detecting a pathogenic antibody versus the potential for false positives. Antibodies have hitherto mostly been tested in serum. However, intrathecal antibody synthesis occurs in most forms of AE and, particularly for NMDA-R antibodies, CSF testing is more sensitive and specific than serum and should be undertaken where clinical suspicion is high and in challenging cases.
Consensus criteria also allow for a diagnosis of ‘seronegative AE’, based on clinical features in the absence of antibodies which may respond to immune therapy (Box 1).6,12
Diagnostic criteria for possible autoimmune encephalitis6
Because of the paraneoplastic associations of several antibodies, investigation for malignancy is crucial. Computed tomography (CT) of the chest, abdomen and pelvis is indicated in all patients with suspected AE. For those with particular associations, such as GABABR, positron-emission tomography is advised and, if negative, should be repeated at 3–6 months.13 Additionally, in NMDA-R encephalitis, because of the association with teratoma, ultrasound of the ovaries or testes should be performed.
Management and outcome
Evidence for optimal management of AE is largely based on retrospective studies, together with principles adapted from other antibody mediated diseases, eg myasthenia gravis. Patients with encephalitis should be managed at a centre with appropriate facilities and specialist expertise, ideally a regional neuroscience centre.
First-line therapy, aimed at reducing antibody levels rapidly, normally comprises intravenous corticosteroids often combined with intravenous immunoglobulin or plasma exchange. In those who fail to respond, more aggressive immune therapy is usually started, with concomitant higher risk of adverse effects, including cyclophosphamide or rituximab.14 In NMDA-R encephalitis, other agents such as bortezomib have been used in a small number of refractory cases. Relapses may occur when immune therapy is reduced, or may reflect tumour recurrence or persistence of missed tumour.15
Starting immune therapy early has been associated with improved outcome in NMDA-R encephalitis in retrospective series. Also, in those who fail to respond to first-line therapy, moving to more aggressive second-line treatments is associated with better outcome and reduced rate of relapse.16 In patients with ovarian teratoma, tumour resection is associated with faster rate of recovery and reduced relapse rate.16
LGI1 encephalitis generally responds well to first-line treatment, particularly corticosteroids, and outcomes are generally good, however, in the long-term cognitive problems are common.17,18
Patients with autoimmune encephalitis often require extensive supportive care, including prolonged intensive care unit stays. Issues may include managing seizures, autonomic instability, infective complications, and agitation. Antipsychotic medications may be required, but should be used with caution due to the risk of extrapyramidal side effects or neuroleptic malignant syndrome, which may be higher in NMDA-R encephalitis.19
With appropriate treatment, outcome overall compares favourably with infective encephalitis, with >80% of patients with NMDA-R encephalitis having no more than slight disability (modified Rankin Scale 2) at 2 years.16 However, there is emerging evidence that subtle neurocognitive/psychosocial issues may be more prevalent than previously appreciated.20 Support from psychology/counselling services and charitable groups, such as the Encephalitis Society, is vital in the recovery phase.
Conclusions
It is increasingly possible to make a positive diagnosis of AE given consensus clinical criteria and increased availability of antibody tests. However, this relies on a high index of suspicion, especially in those presenting with primarily psychiatric features. The diagnosis is often supported by the development of ‘neurological’ features, such as movement disorders and seizures, laboratory tests, and MRI, but these may be normal in some. Antibody testing should be organised in consultation with a neurologist and neuroimmunology laboratory, with a pragmatic strategy based on careful clinical assessment. Even in the absence of antibodies, it may be possible to make a syndromic diagnosis.
Many with AE have a good outcome with early immune therapy, tumour resection if needed, and supportive care. Randomised controlled trials are needed to guide treatment strategies.
Funding
Mark A Ellul receives support from the Association of British Neurologists through a clinical research training fellowship. Benedict D Michael has received funding from the Medical Research Council, Wellcome Trust, British Medical Association, Academy of Medical Sciences and National Institute for Health Research.
- © Royal College of Physicians 2020. All rights reserved.
References








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.