
ABSTRACT
Solitary plasmacytoma is a rare localised neoplasm of monoclonal plasma cells. The standard treatment involves radical radiotherapy; however, a significant proportion of patients subsequently develop multiple myeloma. In this study, we evaluate the outcomes of solitary plasmacytoma in a retrospective cohort of patients treated in a single tertiary centre.
The case records of plasmacytoma patients treated in a 15-year period were analysed and retrospectively followed up from the date of diagnosis. Thirty-four cases met the inclusion criteria; 27 (79%) solitary plasmacytoma of bone (SBP) and 7 (21%) extramedullary plasmacytoma (EMP). The thoracic vertebrae were the commonest sites for SBP while EMP occurred most frequently in the upper airway. Pain and spinal cord compression were the most frequent symptoms. A paraprotein was detectable in 18 (53%) patients. Over a median follow-up of 48 months, 13 (38%) developed multiple myeloma. The 5- and 10-year survival rates were 80% and 56%, respectively; median progression-free survival was 77 months. Four patients (12%) developed a second malignancy.
Progression to multiple myeloma remains a formidable challenge in the management of solitary plasmacytoma, hence adjunct therapies are needed.
Introduction
Solitary plasmacytoma (SP) is a rare localised neoplasm of monoclonal plasma cells presenting as either solitary plasmacytoma of bone (SBP) or extramedullary plasmacytoma (EMP). According to the International Myeloma Working Group (IMWG) criteria, the diagnosis of SP requires a biopsy-proven solitary lesion of bone or soft tissue with evidence of clonal plasma cells, a normal bone marrow with no evidence of clonal plasma cells and an absence of end-organ damage such as hypercalcaemia, renal insufficiency, anaemia or bone lesions (CRAB) attributable to a lymphoplasma cell proliferative disorder.1 Given the growing evidence on the prognostic implication of clonal plasma cells in the bone marrow in SP, a separate diagnostic category of SP with minimal bone marrow involvement (clonal plasma cells less than 10%) has recently been introduced.2–4
SP can be considered part of the spectrum of clonal plasma cell disorders which range from the premalignant monoclonal gammopathy of undetermined significance (MGUS) to multiple myeloma (MM). As with other clonal plasma cell disorders, SP may be associated with a monoclonal protein (paraprotein) detectable in the serum, an excess of kappa or lambda immunoglobulin light chains manifesting as an abnormal serum free light chain (SFLC) ratio and may ultimately progress to MM. The current management of SP focuses on eradication of the primary tumour often with radical radiotherapy or surgical excision, in a minority of cases. While these treatment modalities often result in long-term remissions, a significant proportion of patients experience recurrence or progress to MM. Due to the rarity of this condition, only few prospective studies have been published to date. The evidence base for the current management of SP is mainly limited to relatively small retrospective studies which, nonetheless, have provided insight into the clinical presentation and putative prognostic factors. Bone localisation (SBP), older age (>60 years), persistence of a paraprotein >1 year after therapy and an abnormal serum free light chain ratio at diagnosis have been associated with an increased risk of progression to MM while tumour size <5 cm was reported to be a favourable factor for local control.5–10
In this study, we reported the presentation and outcomes of solitary plasmacytoma in a cohort of patients treated at a large tertiary hospital in the UK. We also aimed to identify risk factors for progression to myeloma and determine the incidence of secondary malignancies in this cohort.
Methods
We retrospectively reviewed case records of adult patients treated between 2002 and 2017 at Addenbrooke’s Hospital, Cambridge. All patients had histological diagnosis and patients with multiple plasmacytomas or coexisting MM were excluded. Clinical, laboratory and radiological parameters at diagnosis and during the course of treatment were analysed until the date of progression to MM and/or death. Proportions were compared using the Chi-squared test. MM-free survival (MMFS) and overall survival (OS) values were derived using the Kaplan–Meier method. Comparison of survival curves was done using the log-rank test. Statistical analyses were performed using Prism version 8.3 (GraphPad, San Diego, USA). The study was performed in accordance with institutional research protocols.
Results
Thirty-four cases of solitary plasmacytoma were identified; 27 (79%) SBP and 7 (21%) EMP. The patient characteristics are summarised in Table 1. The median age at diagnosis was 65 years (range 32–83 years). Two-thirds of patients were male, and this preponderance was more marked for the EMP subset, with 85% being male. The axial skeleton was the commonest site of presentation of SBP; thoracic vertebra (11; 41%), lumbar vertebrae (7; 26%) and cervical vertebrae (2; 7%). The commonest single anatomical site for EMP was in the upper airway (2; 29%) while soft tissue lesions in various anatomical sites (3; 43%). Localised pain, predominantly back pain, was the most frequent presenting symptom of SP in 16 (47%) patients. The next most frequent presenting symptom was spinal cord compression (3; 9%). Other presentations were swelling (2; 6%), pathologic fractures (2; 6%), incidental (1; 3%) and other miscellaneous presentations (10; 29%). A serum paraprotein was detectable in 18 (53%) patients at the time of diagnosis and this was predominantly immunoglobulin (Ig) G in 13 patients (72%) and accompanied by immune paresis (reduction in the uninvolved immunoglobulins) in two patients.
Summary of patient characteristics and treatment outcomes
Twenty-six patients (76%) received radiotherapy alone while two patients received only surgical excision. One patient was diagnosed with metastatic cancer at the same time and did not receive any SP treatment. Most patients (61%) received a total radiation dose of 50 Gy in 25 fractions (range 40–65 Gy). Among the 31 patients who received radiotherapy, the overall response (OR) rate was 84% with a complete response (CR) observed in 24 (77%); an assessment of response was not available for five patients. Local relapse occurred in two (7%) patients. The radiation dose was at least 50 Gy in both.
At a median follow-up of 48 months, 13 (39%) patients had progression to MM. The median time to progression was 22 months but, notably, two male patients had progression more than 5 years after radiotherapy (5.2 and 7 years). Compared with the no-progression group, a higher but statistically non-significant proportion of male patients experienced progression to MM (85% vs 57%; p=0.09). Similarly, 50% of patients with a non-IgG paraprotein developed MM compared with 18% in the no-progression group (p=0.15).
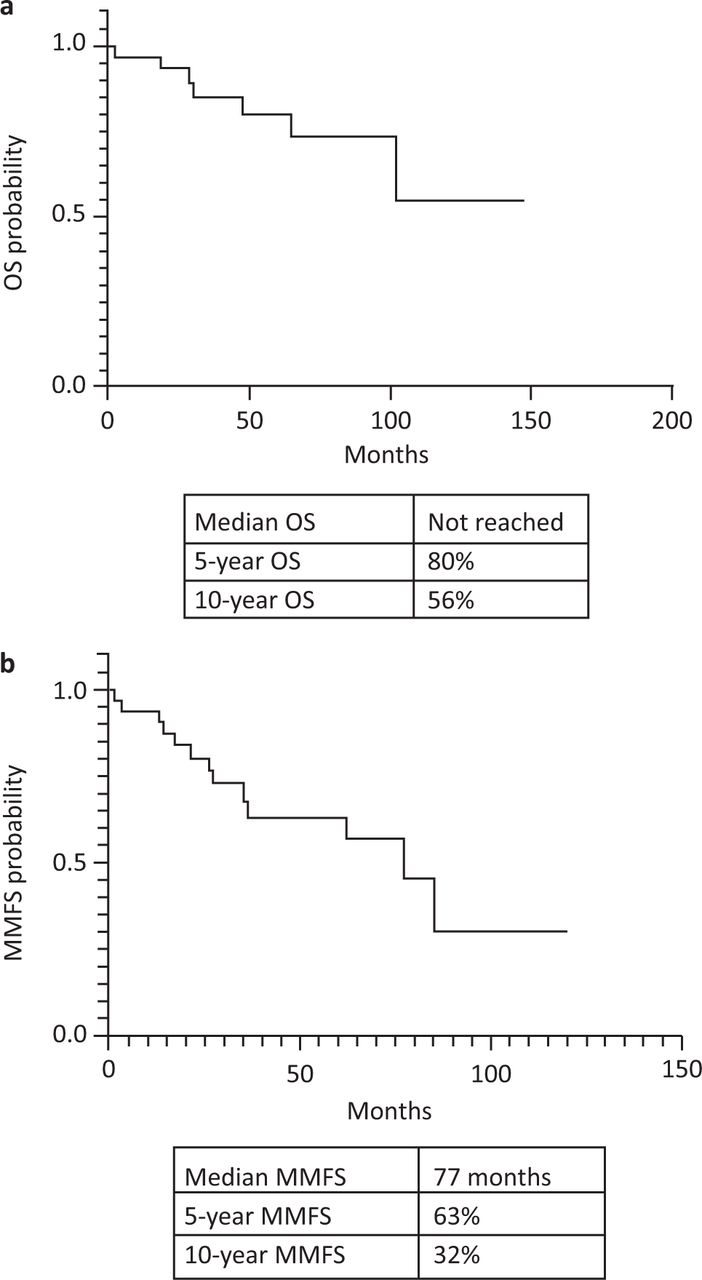
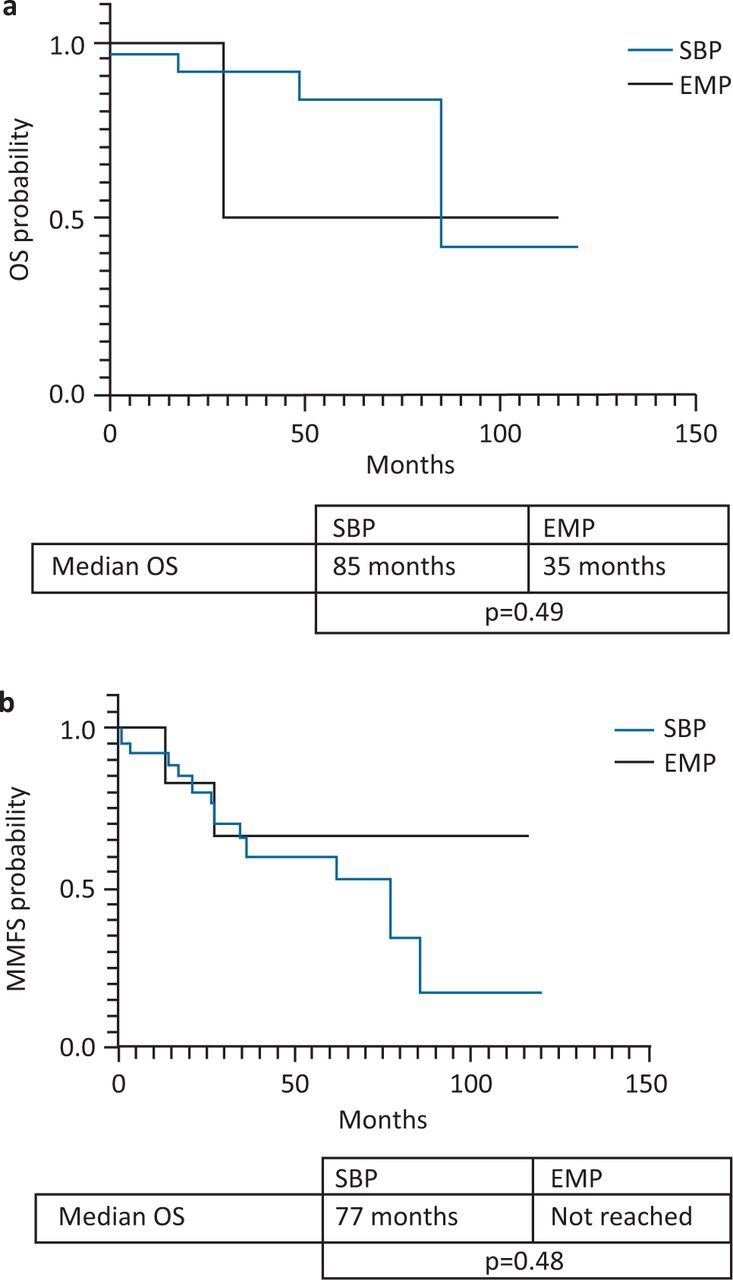
The 5- and 10-year OS were 80% and 56%, respectively, for the entire cohort (Fig 1a). The median MMFS was 77 months and 5- and 10-year MMFS were 63% and 32%, respectively (Fig 1b). The median overall survival for SBP and EMP were 85 months and 35 months, respectively (Fig 2a) but this difference was not statistically significant (p=0.49). The SBP group had an inferior, but statistically non-significant median MMFS survival compared with the EMP group (77 months vs not reached; p=0.48; Fig 2b).
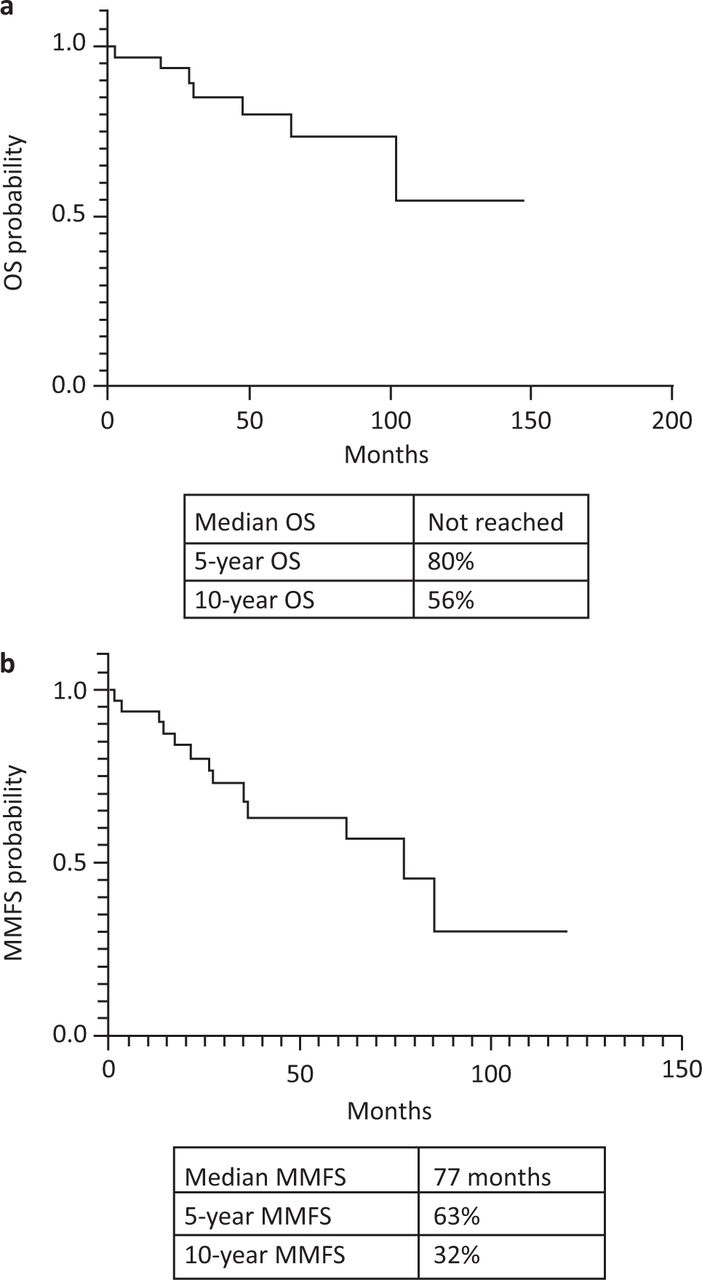
All patient survival. a) Overall survival (OS). b) Multiple myeloma-free survival (MMFS).

Solitary plasmacytoma of bone (SBP) versus extramedullary plasmacytoma (EMP). a) Overall survival (OS). b) Multiple myeloma-free survival (MMFS).
At 170 person-year follow-up, four (12%) patients developed a second malignancy; two lung cancer, one diffuse large B cell lymphoma (DLBCL) and one chronic myelomonocytic leukaemia (CMML). The median time to diagnosis of the second malignancy was 4 years (range 3–5 years). Interestingly, three of the four patients developing a second cancer had EMP.
Discussion
The patterns of presentation of SP in our cohort were similar to previous studies with respect to age, sex and localisation with the majority of SBP presenting in the thoracolumbar spine and EMP in the soft tissues of the head and neck.11
Radical radiotherapy has been the cornerstone of the management of SP, offering excellent results in the majority of patients. However, progression to MM remains a significant challenge. Currently, there are no validated biomarkers which could prospectively identify the group of patients with a high risk of progression. Previous studies have suggested advanced age (>60 years), abnormal serum free light chains and persistence of paraprotein after radiotherapy as high-risk factors.8,9,12 In our study, while statistical significance was not achieved due to the small sample size, higher proportions of male patients and those with non-IgG paraprotein were observed in the high-risk category. Interestingly, the presence of a non-IgG paraprotein has been shown to predict a high risk of MGUS progression to MM but this association has not previously been described in SP and therefore warrants further investigation in larger series.13 Furthermore, the two patients in our cohort who had immune paresis progressed to MM within 17 months and 21 months, indicating a possible association with high-risk disease. However, we recognise that the small sample size of this study limits our ability to make firm conclusions about these findings.
SBP has been consistently shown to predict an increased likelihood of progression to myeloma but there have been conflicting results about the impact of SP type on overall survival. In this study, likely due to the small sample size, we found no difference in overall survival between SBP and EMP but there was a trend towards inferior myeloma-free survival for patients with SBP although statistical significance was also not reached. It is noted that unlike the SBP patients, all the three EMP patients progressing to MM in our cohort occurred prior to 2010, before novel myeloma therapies became widely available on the NHS and this might have confounded the OS results since death often occurs as a result of progression to MM. However, our OS results were similar to the findings of Barzenje et al in a retrospective study of 77 SP patients with longer follow up.14 A similar-sized study by Suh et al reported results consistent with ours despite the inclusion of patients who received myeloma-directed chemotherapy.15 A large US population-based study of 2,785 SP patients reported inferior OS and progression-free survival (PFS) for SBP, findings which were similar to the study by Finsinger et al.12,16 A possible explanation for the differences in outcomes between studies could be the recent improvement in myeloma treatment with the introduction of novel agents. Stringent diagnosis of SP might have also had a significant impact on SP survival as demonstrated in the findings of a recent publication by Sharpley et al of a cohort of SP patients treated in the UK and Brazil.17 The 5-year OS of 97.5% was significantly higher than in our cohort and other previous studies but the median PFS of 61 months was comparable. The reason suggested by the authors include the increased use of sensitive radiological imaging such as whole-body computed tomography / fluorodeoxyglucose positron emission tomography to correctly identify SP thereby excluding high-risk multifocal plasmacytoma/myeloma cases from the cohort. Furthermore, some of the patients treated in our cohort were diagnosed at a time when sensitive tests such as multiparameter flow cytometry, serum free light chain assay and magnetic resonance imaging were not widely in use in this setting. These investigations serve to identify patients with occult multifocal/systemic disease who are now offered myeloma-directed chemotherapy rather than radiotherapy alone.
The optimal radiation dose for the treatment of SP has historically been a contentious issue among radiation oncologists with a range of doses favoured by different hospitals. In our centre, the current standard regimen is 50 Gy delivered over 25 fractions for tumours >5 cm and 40 Gy in 20 fractions for tumours <5 cm; however, some patients treated earlier received varying doses. There is now considerable evidence from the literature that higher radiation doses confer no additional benefit over standard doses. Knobel et al found no dose-response relationship for radiation doses greater than 30 Gy, even for larger tumours; of the 201 patients with SP who received radiotherapy in this study, the local failure rate for patients who received 30 Gy was 7% compared with a rate of 12% for those who received >30 Gy.7 Another study of 46 patients receiving radiotherapy for SP similarly found no association between radiation dose and local failure or progression to MM but found higher local failure rates for bulky tumours (>5 cm) treated with radiation doses <35 Gy.10 Overall, it appears that a total radiation dose of 50 Gy appears optimal for local control in most cases of SP as only two local relapses (6%) were identified in our cohort. This is in agreement with the recently published European Expert Panel recommendation of 40–50 Gy delivered over approximately 4 weeks.18 Surgery remains a viable modality of treatment in cases of EMP where complete surgical excision is possible or in anatomical locations where radiotherapy would be associated with unacceptable side effects.
A role for adjuvant chemotherapy has been suggested in the management of SP but there is limited evidence as only few prospective clinical trials have been published. A randomised trial of low-dose melphalan and prednisolone following radiotherapy for SBP versus radiotherapy in 53 patients with SBP reported significant improvement in disease-free survival for the combination treatment group.19 Interestingly, no increase in secondary haematological malignancies was reported despite prolonged administration of an alkylating agent. Nevertheless, since this publication, the treatment of myeloma has experienced significant developments with the introduction of immunomodulatory agents and proteasome inhibitors. The role of these new drugs as adjuvant therapies in SP has not been determined as there are no published randomised clinical trials. However, an ongoing clinical trial of the combination of lenalidomide and dexamethasone in high-risk SBP patients will hopefully shed more light on the role of these agents and the results are eagerly awaited.20
As observed in our cohort, the majority of MM progressions occurred within 5 years of radiotherapy, however, given the two late relapses in our cohort and similar late relapses in a larger cohort, even after 10 years, we recommend lifelong monitoring for evidence of MM.7 Furthermore, while previous studies have suggested an increased incidence of both haematological and solid cancers in myeloma patients, this relationship had not previously been extensively investigated in SP.21–23 Barzenje et al reported the development of a second malignancy in 14% of their SP cohort.14 In this study, over the course of follow up of 170 person-years, there were four (12%) cases of new malignancies with lung cancer being the most frequent giving an overall cancer incidence rate of 30 per 1,000 person-years. The median time from SP diagnosis to diagnosis of second cancer was 4.5 years. Of the two patients who developed lung cancer, only one received radiotherapy to the thoracic spine. The other patient had EMP localised to the jejunum which had been surgically resected. While it is difficult to exclude the influence of other risk factors in this small retrospective cohort, the incidence of secondary cancers in SP requires further investigation in larger studies.
Conclusion
The diagnosis of SP should always be considered in the differential diagnosis of patients presenting with solitary bone lesions. Appropriate biopsies should be arranged without delay as prompt histological diagnosis and treatment with radical radiotherapy offers excellent local control and long-term remissions for the majority of patients with SP. However, there remains an unmet need for novel adjunct therapies to prevent or delay progression to MM in high-risk groups as well as validated biomarkers to identify patients who will require additional treatment. Based on our data, we recommend long-term monitoring of SP patients for evidence of progression to MM or the development of other malignancies.
Acknowledgements
The authors wish to express their immense gratitude to Dr Jenny Craig for her review of the initial draft of this article.
- © Royal College of Physicians 2020. All rights reserved.
References








{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.