
ABSTRACT
Breathlessness is a subjective symptom that may stem from a number of pathological and functional aetiologies. Consequently, clinicians are often faced with the challenge of navigating between the tensions of Occam's razor (parsimonious aetiology) or Hickam's dictum (multiple diagnoses). We report a case of a 36-year-old woman with a lifelong history of episodic breathlessness caused at various times by dysfunctions of lung parenchyma (emphysema) and airway smooth muscle (asthma), skeletal muscle (filamin-C fibrillary myopathy) and cardiac muscle (cardiomyopathy). We illustrate the utility of the modern diagnostic toolbox in the assessment, understanding and management of complex dyspnoea (including the use of inflammometry, inhaled-gas magnetic resonance imaging-guided bronchial thermoplasty, and genetic testing), and also demonstrate the importance of interdisciplinary data interpretation in establishing accurate aetiologic diagnoses.
Case presentation
A 36-year-old woman, who never smoked, presented for assessment of severe asthma and breathlessness. Born prematurely at 26 weeks, she was diagnosed with asthma at the age of 3 years old. Other medical history included hypogammaglobulinaemia, an unprovoked pulmonary embolism, hypothyroidism, insulin-dependent diabetes mellitus, possible Gilbert's syndrome and fibromyalgia.
At the time of her assessment, in addition to 10 mg prednisone, she was on daily fluticasone/salmeterol (1,000/100 μg), ciclesonide 400 μg, tiotropium 18 μg and montelukast 10 mg, though she remained symptomatic (Asthma Control Questionnaire-7 (ACQ-7) = 2.0; Asthma Quality of Life Questionnaire (AQLQ) = 5.8). Her blood and sputum cell counts and fractional exhaled nitric oxide (FeNO) were normal on two occasions, including at the time of an exacerbation. Peak flow variability recorded over 2 weeks was >30%. Her forced expiratory volume in 1 second (FEV1) was 0.76 L (26% predicted) and improved to 1.30 L after inhaling salbutamol. Her forced vital capacity (FVC) was 1.92 L (57% predicted) and improved to 2.57 L after inhaling salbutamol. Thoracic computed tomography revealed moderate pan-lobular tissue attenuation which reflected terminal airspace enlargement that had been attributed to neonatal respiratory distress syndrome.
Given her variable airway obstruction without evidence of airway inflammation, we attributed her persistent symptoms to smooth muscle dysfunction. We performed inhaled-gas magnetic resonance imaging (MRI) which revealed substantial focal ventilation defects throughout both lungs, some of which improved following salbutamol inhalation (Fig 1).1,2 She underwent a single session of MRI-guided bronchial thermoplasty, treating only those airways that were proximal to salbutamol-responsive ventilation defects.2 Since benefits of thermoplasty are best when performed in those patients without airway inflammation, the procedure was performed after normalising sputum cell counts and FeNO.3 Over the next 2 years, her asthma control and quality-of-life improved (ACQ-7 = 1.6; AQLQ = 6.0) and she was weaned off prednisone without exacerbation. Inhaled-gas MRI showed ventilation improvements in lung regions distal to thermoplasty treated airways. Baseline FEV1 improved to 0.97 L (35% predicted) and increased to 1.36L (50% predicted) post-salbutamol.
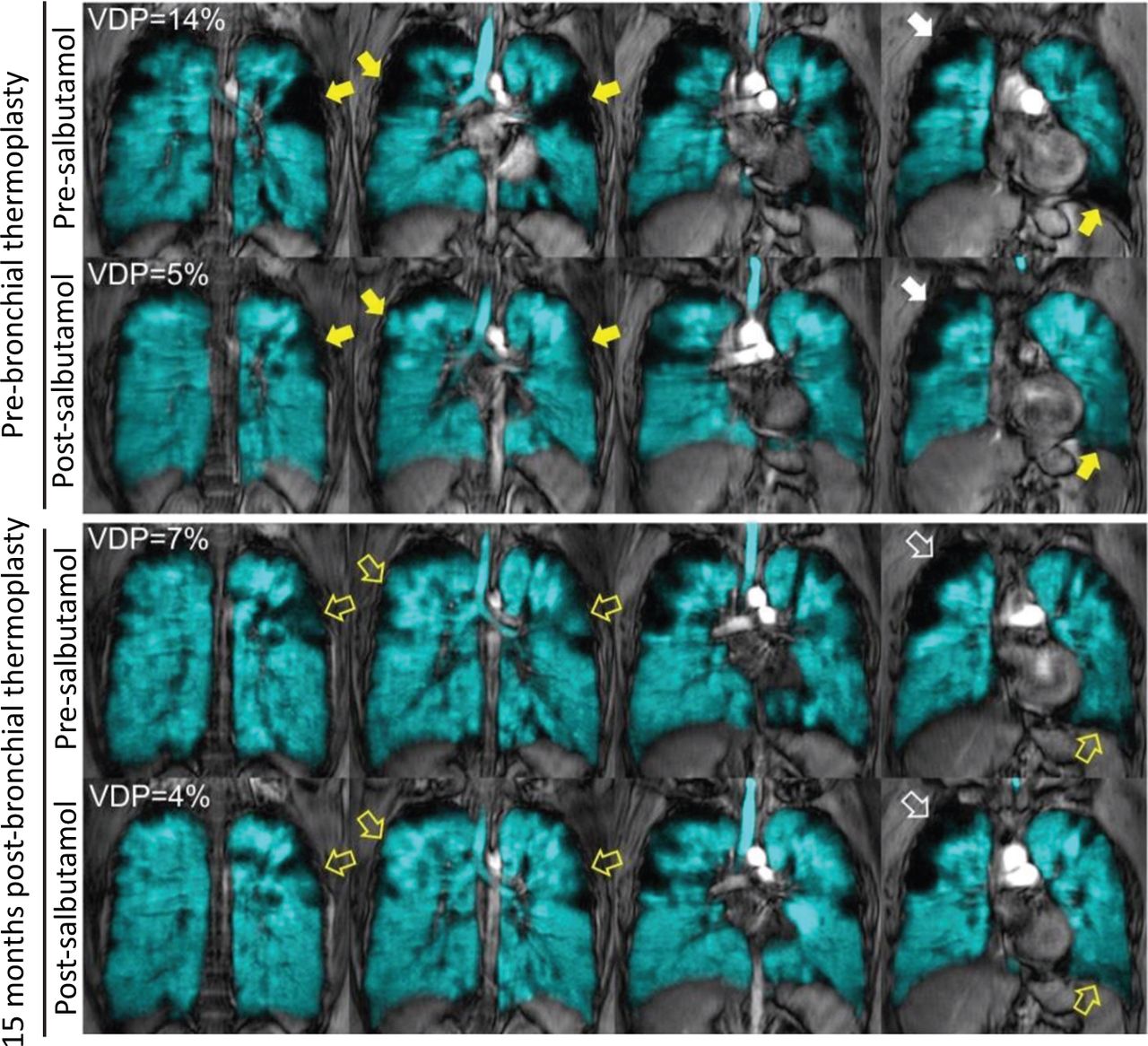
Inhaled hyperpolarised 3He magnetic resonance imaging pre- and post-salbutamol, acquired before and 15 months after a single session of magnetic resonance imaging-guided bronchial thermoplasty. 3He gas distribution (in cyan) co-registered to the 1H of the thorax (in grey scale) is shown for the four centre coronal slices acquired at each timepoint. Before bronchial thermoplasty, abnormal ventilation was evident throughout the lung. Focal ventilation defects that improved (yellow solid arrows), other defects persisted (white solid arrows). Fifteen months following bronchial thermoplasty, pre-salbutamol VDP was improved to 7%; visually improved ventilation in the regions treated by bronchial thermoplasty (yellow bordered arrows). Focal ventilation defects persisted following salbutamol inhalation prior to bronchial thermoplasty (white bordered arrows). VDP = whole lung ventilation defect percent.
She presented again 2 years later with worsening breathlessness, despite stable lung function. She reported mild dysphagia, fatigue and shoulder girdle weakness. She had developed nocturnal alveolar hypoventilation that required overnight volume-assured pressure support. Serum creatine kinase was mildly elevated. Electromyography showed small-amplitude motor unit potentials with evidence of polyphasia and early recruitment. A deltoid muscle biopsy revealed septal perivascular and endomysial chronic inflammation and type 2 fibre atrophy. There were no myofibrillar changes.
Given this clinical presentation, and knowledge that her son had mild hypotonia, she underwent genetic testing of 94 genes associated with muscle disorders.4 Pathogenic variants were detected in two genes: FLNC (NM_001458.4: c.7371delT; p.Ser2457fs) and CHRNE gene (NM_000080.3: c.448C>T; p.Ser163Leu). A diagnosis of filamin-C-related fibrillary myopathy was determined responsible for her myopathy based on the presence of the FLNC variation, which is known to cause a frameshift and introduce a premature stop codon in exon 44, with resultant nonsense-mediated messenger ribonucleic acid decay and haplo-insufficiency. She did not have features of congenital myasthenic syndrome and only one variant of uncertain significance was found in the CHRNE gene, a bi-allelic disorder. She was recommended to continue regular exercise together with creatine monohydrate and coenzyme Q10 supplementation.
Her dyspnoea continued to worsen, and in 2019 she was diagnosed with heart failure. Cardiac MRI showed left ventricular ejection fraction of 29% and mild positive late gadolinium enhancement in the mid-myocardium, in a pattern consistent with muscle dystrophy. Aggressive therapy with spironolactone, bisoprolol, sacubitril-valsartan and ivabradine improved her symptoms and her N-terminal pro B-type natriuretic peptide decreased to near normal (449 pg/mL from 1,011 pg/mL, initially). Right heart catheterisation 3 months later showed a normal cardiac output and filling pressures.
Discussion
We report the evolution of symptoms in a young woman with complex medical conditions. Her case highlights an important clinical message: breathlessness can be multifactorial and, in this patient, dysfunctions of smooth muscle (asthma), skeletal muscle (filamin-C myofibrillar myopathy) and cardiac muscle (cardiomyopathy) evolved and contributed to her dyspnoea. Moreover, to our knowledge, this is the first report of a patient in whom asthma and myofibrillar myopathy coincide, which demonstrates the growing utility of gene panel-sequencing in medical diagnosis. In clinical medicine, it is common practice to come up with parsimonious explanations for disease symptoms. Breathlessness is a symptom where the Occam's razor principle may not be helpful. This patient had a multisystem impairment that evolved over time, all contributing to her dyspnoea; an illustration of Hickam's dictum.
Nevertheless, we considered a potentially unifying explanation for the evolution of her disease. Heterozygous mutations of the FLNC gene cause autosomal dominant filamin-C-associated forms of myofibrillar myopathy, a class of protein aggregate myopathies defined by the presence of desmin-positive protein aggregates and degenerative inter-myofibrillar network changes.5,6 The gene is expressed in both skeletal and cardiac muscle cells. Functional mutations in the FLNC actin-binding domain can confer a progressive adult-onset distal myopathy phenotype or a mild mixed proximal and distal phenotype (myofibrillar myopathy), and variants in this gene have also been associated with cardiomyopathies.7,8 Although the heterozygous variation c.7371delT has been previously reported in the ClinVar database, its clinical implications are unknown. It appears unlikely that the FLNC gene mutation explains her asthma; although the gene is expressed in both airway and vascular smooth muscle cells, the loss of function mutation observed in our patient does not explain increased airway smooth muscle contractility.9
In summary, we report on the evolution of multifactorial breathlessness in a patient who subsequently developed a myopathy and cardiomyopathy due to a mutation of the FLNC gene, all of which was revealed using an interdisciplinary asthma management strategy including inflammometry and inhaled-gas MRI-guided bronchial thermoplasty.10
Funding
Parameswaran Nair was supported by a Frederick E Hargreave Teva Innovation Chair in Airway Diseases. Carmen Venegas was supported by a Pontificia Universidad Católica de Chile fellowship. Sarah Svenningsen was supported by Canadian Institutes of Health Research (CIHR) Banting and Canadian Respiratory Research Network (CRRN) post-doctoral fellowship awards. Grace Parraga holds a tier 1 Canada research chair in lung imaging, funded by the Canada Research Chair programme.
Conflicts of interest
Parameswaran Nair has received grants and personal fees from AstraZeneca, Novartis, Teva and Roche; grants from Sanofi; and personal fees from Merck and Equillium outside of the submitted work.
- © Royal College of Physicians 2020. All rights reserved.








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.