
Sub-acute progressive muscle pain and weakness
A 58-year-old woman was transferred to a tertiary neurology unit with a 10-week history of progressive symmetrical proximal muscle weakness with associated myalgia, predominantly affecting the lower limbs. She had no visual disturbances, speech difficulties, hearing loss, dysphagia, fasciculation, sensory deficit, bowel nor bladder dysfunction, cognitive impairment, shortness of breath, rash, arthralgia or episodes of dark urine.
She had a history of type 2 diabetes mellitus, lichen sclerosis and a single convulsive seizure 1 year prior to presentation that was not investigated at the time. Her regular medications included metformin, humulin M3 and atorvastatin 20 mg, which she had been taking for approximately 5 years. Her general practitioner had discontinued her atorvastatin 2 weeks prior to hospital admission but her weakness continued to progress rendering the patient bed bound.
Initial investigations demonstrated elevated serum creatine kinase (CK) at 2,979 U/L (reference range 25–200), alanine aminotransferase (ALT) at 811 U/L (reference range 0–40) and aspartate transaminase (AST) at 445 U/L (reference range 0–40) with normal bilirubin, alkaline phosphatase and gamma glutamyl transferase levels. Suspecting a diagnosis of myositis, a biopsy of the deltoid muscle was performed, which instead showed an active necrotising myopathy characterised by several acutely necrotic muscle fibres, scattered regenerating muscle fibres, patchy sarcoplasmic major histocompatibility complex class 1 (MHC-1) up-regulation and membrane attack complex (MAC; complement 5b-9) deposition (Fig 1). Serology was positive for anti-3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR) antibodies while other myositis-specific autoantibodies were negative, consistent with a diagnosis of statin-induced immune-mediated anti-HMGCR necrotising myopathy. Radiological screening for malignancy was negative.
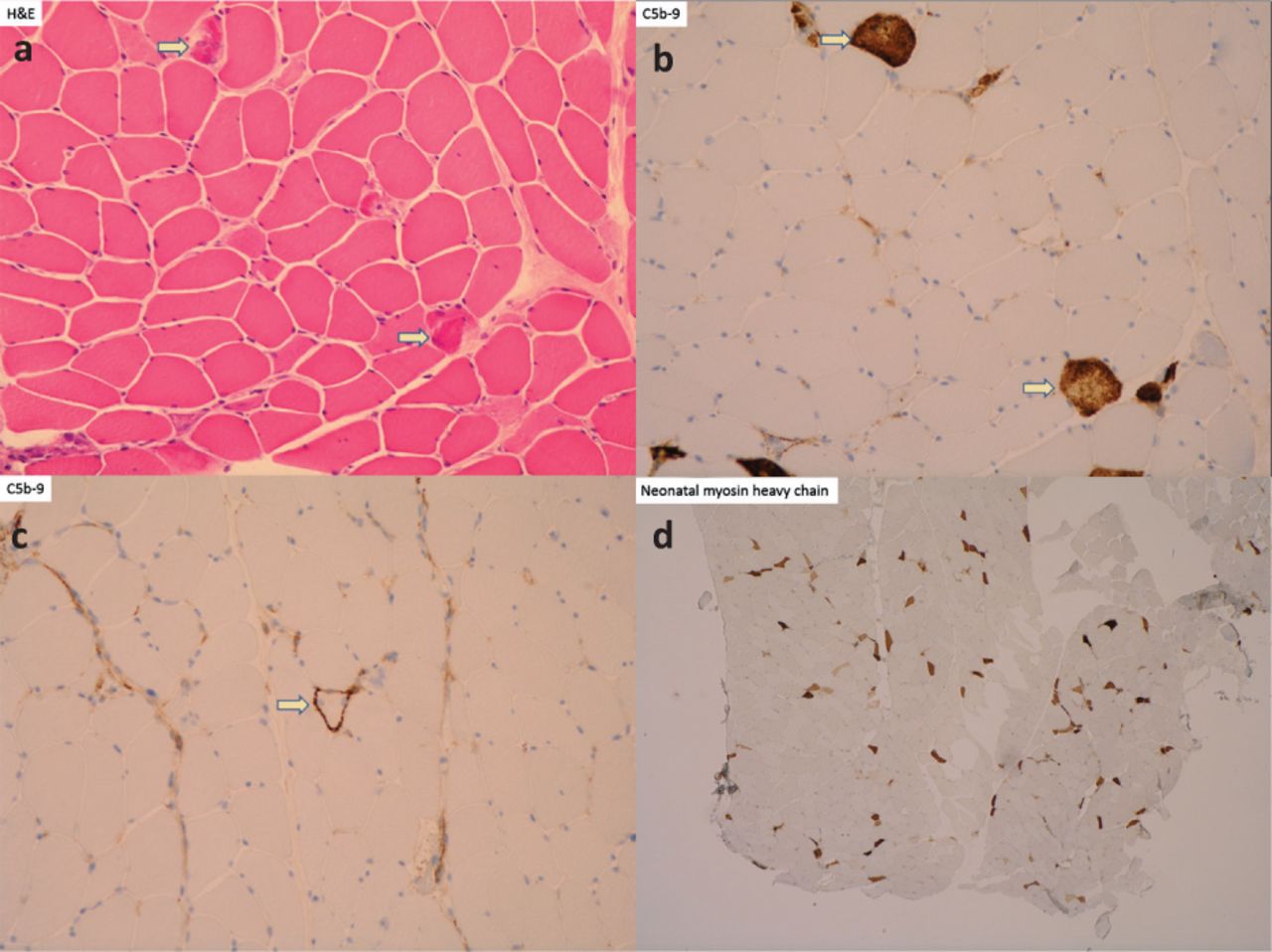
Muscle biopsy histopathology slides. a) Hematoxylin and eosin showing acutely necrotic muscle fibres. b) Complement 5b-9 (C5b-9) stain showing strong sarcolemmal labelling among necrotic fibres. c) C5b-9 stain showing pre-necrotic fibres. d) Neonatal myosin heavy chain stain showing numerous, likely regenerating muscle fibres.
The patient was started on pulsed intravenous methylprednisolone 1 g for 5 days, followed by high-dose oral prednisolone 60 mg. After 2 weeks of steroid therapy and no symptom improvement, she was administered a 5-day course of intravenous immunoglobulins (IVIg). Later commenced on mycophenolate mofetil (MMF) as a corticosteroid-sparing agent and established a prednisolone tapering regimen. CK levels improved to 599 U/L. With negligible improvement of her symptoms, she was transferred to a neuro-rehabilitation centre and discharged home 2 months later.
At 3-month follow-up she was taking prednisolone 30 mg and MMF 2 g. The patient still had profound proximal muscle weakness and required a wheelchair for mobilisation.
What is immune-mediated necrotising myopathy?
Statins are the most commonly prescribed cholesterol-lowering agents worldwide. They reduce cardiovascular morbidity and mortality through inhibition of HMGCR, a key regulatory enzyme in cholesterol biosynthesis. Although statins are often well tolerated, these drugs can induce a spectrum of myopathies, ranging from asymptomatic elevation of CK and myalgia to rhabdomyolysis and immune-mediated necrotising myopathy (IMNM).
Statin-induced IMNM is a rare and relatively novel disease that may be mistaken clinically for an idiopathic inflammatory myopathy (such as polymyositis, dermatomyositis or inclusion body myositis).1 It is characterised by severe symmetrical proximal muscle weakness with or without myalgia, significantly elevated CK, myofibre necrosis with minimal or absent inflammatory cell infiltration on muscle biopsy, and positive anti-HMGCR antibodies.1–2 In addition to CK, other muscle-derived enzymes may be elevated (including ALT and AST), which are more commonly associated with hepatic cell injury. Any patient presenting with persistent myopathy after statin withdrawal should prompt a suspicion of anti-HMGCR necrotising myopathy. Patients may have been taking a statin for several months or years before they develop anti-HMGCR antibodies.1 There is an increased, albeit low, risk of malignancy among these patients, thus, screening is imperative.2
What are the differential diagnoses?
The differential diagnoses for a sub-acute symmetrical upper and lower limb myopathy is wide and includes rhabdomyolysis; inflammatory myositis; toxin- and drug-induced myopathy; late onset congenital or metabolic myopathy; and adult onset spinal muscular atrophy. A neuromuscular junction disorder should be considered, however, a lack of fatigability would be atypical for myasthenia gravis. Anterior horn cell disease secondary to acute poliomyelitis can involve all limbs but typically presents with asymmetric weakness, furthermore it is considered eradicated within the UK.
Why is this case important?
Discontinuation of the offending statin is the foremost important treatment but the vast majority of patients will require aggressive immunosuppressive therapy to prevent long term disability.3 Current consensus is to treat empirically with corticosteroids; IVIg is also recommended as a first-line therapy.2 The addition of a second-line immunosuppressive agent (such as azathioprine, methotrexate or MMF) is often required.3 A significant proportion of patients have persistently active disease despite aggressive immunosuppression. Several case reports have suggested a role for rituximab, a chimeric monoclonal antibody targeting CD20-expressing B cells, in the management of refractory anti-HMGCR necrotising myopathy.4
Acknowledgements
We would like to thank Dr Tuomo Polvikoski, consultant neuropathologist, for provision and analysis of the histopathological images.
- © Royal College of Physicians 2021. All rights reserved.








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.