
ABSTRACT
Interstitial lung diseases (ILDs) are a heterogeneous group of diseases characterised by varying degrees of fibrotic and/or inflammatory abnormalities of the lung parenchyma. Management of ILD is often challenging for non-respiratory physicians. We discuss the respiratory assessment and management of patients with ILD presenting with acute breathlessness on the acute take, including acute exacerbations of ILD.
Key points
There are seven main supervening respiratory diagnoses that may lead to decompensation in interstitial lung disease (ILD) patients.
An acute exacerbation of an ILD is a clinical diagnosis of exclusion. Treatment is challenging due to a lack of high-quality clinical trial evidence.
Antibiotics should be considered in all exacerbating patients, at least initially, particularly in those who are immunosuppressed.
Patients with idiopathic pulmonary fibrosis or other types of advanced fibrotic ILD who exacerbate generally have poor outcomes. Mainstay treatment for these patients is usually supportive care rather than escalation to intensive care.
Patients with inflammatory ILDs who exacerbate should be assessed for treatment with high-dose immunosuppression.
Introduction
The interstitial lung diseases (ILDs) are a heterogeneous group of more than 200 conditions characterised by varying degrees of fibrotic or inflammatory abnormalities of the lung, or a combination of the two. Untreated, some may progress to respiratory failure and death over varying timescales but, if appropriately treated, the more inflammatory and mixed phenotypes can fare better overall. Although there are a large number of ILDs, in usual clinical practice, a smaller number are more commonly seen on the acute take (Table 1; Fig 1). Some drugs may also cause ILD (Table 2).
Brief overview of interstitial lung diseases that might present to the acute take
Common respiratory causes of respiratory failure in the patient with ILD presenting with breathlessness to the acute take with suggested investigations and management advice
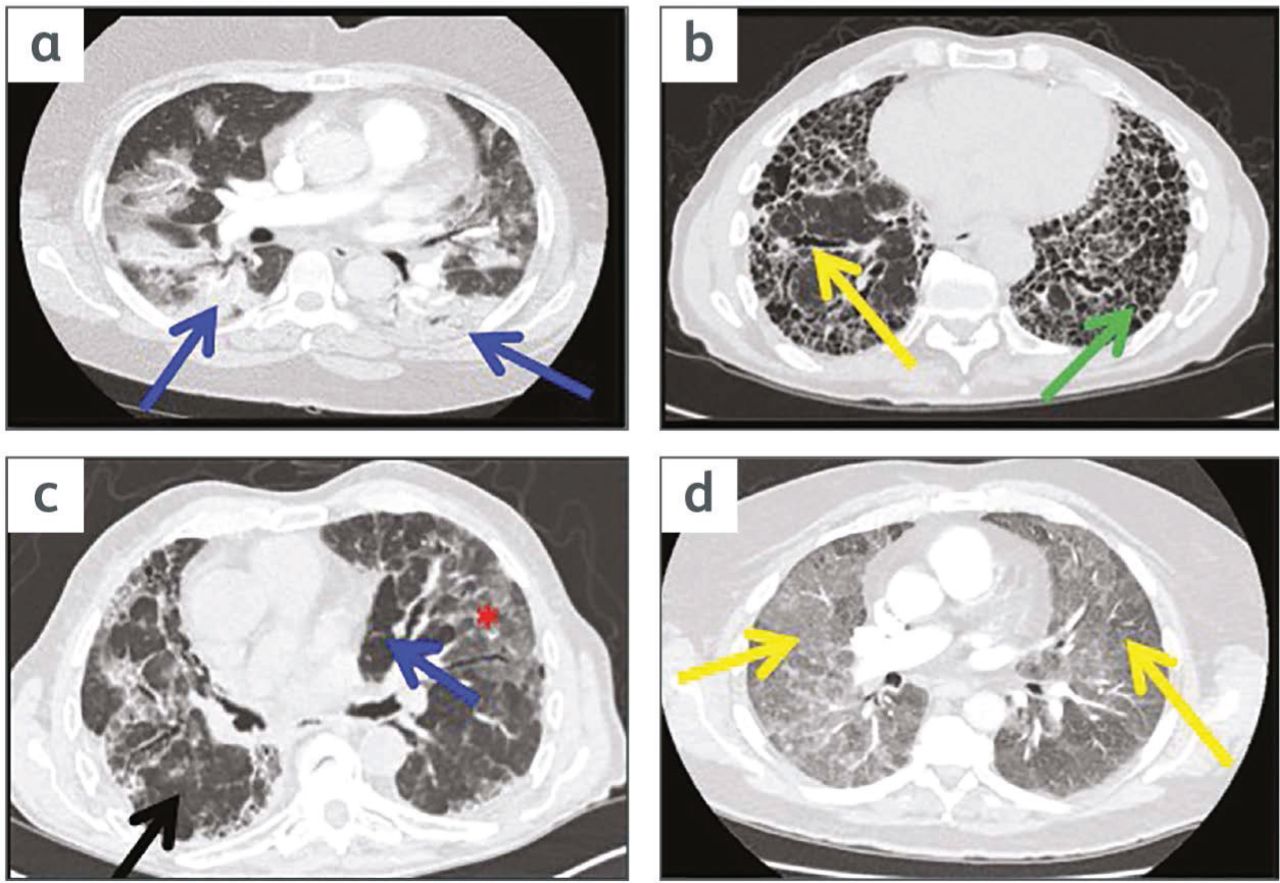
High-resolution computed tomography (HRCT) images of some common fibrotic and inflammatory interstitial lung disease (ILD) patterns that might be seen on the acute take. a) A patient with an inflammatory type of ILD - organising pneumonia (OP) - showing bilateral consolidation (blue arrows). b) A patient with fibrotic type of ILD - idiopathic pulmonary fibrosis. Features are honeycombing (green arrow), traction bronchiectasis (yellow arrow) and reticulation. c) A patient with fibrotic hypersensitivity pneumonitis. It demonstrates ‘mosaic perfusion’ with areas of high attenuation or ground glass opacity (red asterisk) and lucent or dark areas (blue arrow) and areas of normal lung (black arrow). If juxtaposed like this the three areas of differing density are known as the ‘headcheese sign’. d) A patient with inflammatory hypersensitivity pneumonitis. This shows extensive ground-glass opacity and fluffy centrilobular nodules (yellow arrows).
Treatment plans need to be tailored to the specific disease entity. Diagnosing and treating ILD can be clinically challenging, and a multidisciplinary team (MDT) approach is recommended to determine the most accurate diagnosis and appropriate management plan. An ILD MDT should ideally consist of consultants, preferably with experience in ILD, from respiratory medicine, thoracic radiology and pathology. The latter is only needed if lung tissue biopsy material is available for review. However, it is recognised that not all hospitals will have the workforce or infrastructure for an ILD MDT at present. Specialist ILD MDT advice should be available, however, from a specialist ILD centre in each region of the UK.
Many of the ILDs are uncommon or rare and the majority of ILD work is done via respiratory outpatient clinics. This leaves little opportunity for the non-respiratory specialist to gain significant exposure or experience in their diagnosis and management. Yet these patients will often present acutely unwell and, sometimes, in respiratory failure on the acute unselected medical take. The purpose of this article is to give an overview of the approach to the diagnosis and management of the hypoxaemic patient with ILD presenting in this setting.
Management of the hypoxaemic patient with ILD presenting to the acute medical take
There are two likely scenarios that the non-respiratory physician may meet:
acute respiratory failure in a patient already known to have a pre-existing diagnosis of an ILD
acute respiratory failure in a patient with a likely or suspected de novo presentation of an ILD.
The ‘de novo’ presentation is often more difficult to manage because the lack of a pre-existing established ILD diagnosis can make treatment decisions and judging prognosis harder. It is important to assess whether the ILD is predominantly fibrotic or inflammatory in nature and how severe the disease is, as these factors will influence management and prognosis.
Tip
Patients with a primary respiratory diagnosis of ILD without other significant respiratory decompensating comorbidities will usually present in type I respiratory failure (RF) rather than hypercapnic type II RF. Clinicians should be wary of the known patient with ILD who presents in type II RF. They will likely either have a secondary factor contributing to their RF; for example, obesity, co-existing airways disease (asthma or chronic obstructive pulmonary disease (COPD)) or fluid overload / heart failure, which are additional diagnoses that may need treatment in their own right or they may be approaching end of life.
Establishing the cause and managing the respiratory deterioration
There are seven main respiratory-related diagnoses that may cause decompensation in patients with ILD (Table 2). Obtaining a good clinical history and thorough physical examination will guide the clinician towards the most likely scenario. There are of course many other non-respiratory causes of increased breathlessness that may also need to be considered but these will not be covered here.
Infection / opportunistic infection in the patient with ILD
Diagnosing and managing a ‘chest infection’ is bread-and-butter work for the acute take. There are a few ILD specific points to consider, however, in patients with one of the commonest ILDs, idiopathic pulmonary fibrosis (IPF), who may be taking anti-fibrotic (AF) drugs (pirfenidone or nintedanib), and also in patients with other ILDs who may be taking various immunosuppressive (IS) regimens for inflammatory ILD.
The AF drugs do not suppress the immune system to any significant extent. They do not need to be stopped in the presence of a concurrent infection. There may be other reasons, however, to consider stopping them in an acutely unwell patient. These include derangement of liver function tests, when creatinine clearance is below 30 mL/min eg in an acute kidney injury setting (risk of increased drug toxicity), and/or in the presence of nausea, vomiting, ongoing weight loss (known side effects of AF drugs) or because the patient is nil by mouth. There is no intravenous equivalent for these drugs and no pressing need to give them via an nasogastric tube, even if there is one in situ. Pausing them does not lead to rebound disease progression.1,2 To help decide when it is best to restart these drugs peri-hospitalisation, please seek advice from the prescribing specialist ILD centre. To maintain safety, AF drugs require liver function test monitoring and an appropriate time to recheck these after a drug re-start can then be put in place.
Various immunosuppressive (IS) drug regimens are used in the management of the different types of inflammatory ILD (eg in connective tissue disease (CTD) - ILD). The majority of patients will be on various doses of oral corticosteroid monotherapy, second-line IS drug monotherapy (such as mycophenolate mofetil (MMF), methotrexate (MTX), azathioprine (AZA)) or dual drug combinations of corticosteroid and a second-line IS drug.
The management of the acutely unwell patient on long-term corticosteroids is well established. The corticosteroid should not be stopped suddenly, even when there is concurrent infection, and often doses are increased to prevent an adrenal crisis. Second-line IS drugs prescribed for ILD management, however, are usually stopped in the setting of an acute infection, particularly when the infection has been severe enough to precipitate a hospital admission. Some IS drugs (such as MTX) have a long half-life and so will continue to influence the lung disease for some time after discontinuation. However, other drugs have much shorter half-lives (such as MMF and AZA). In the setting of significant infection, the risk-benefit analysis will almost always fall in favour of stopping IS drugs.
Patients with CTD-ILD may be on more complex IS regimens and some of their drugs may be aimed at the management of their joint disease rather than their lungs. It is usually safe to temporarily stop these drugs on the first day or night of the patient's admission. Early subsequent advice can then be sought from specialist respiratory or rheumatology teams regarding ongoing drug management.
For patients on higher doses of IS drugs, opportunistic infection needs to be considered. Most at risk are those taking high-dose corticosteroids (>40 mg/day prednisolone), those on steroid-immunosuppressive dual drug combinations or those who have recently received even more powerful immunosuppressive drugs such as intravenous cyclophosphamide typically for a CTD-ILD.
Pneumocystis jirovecii (PJP) is one such infection to consider. The radiological pattern of PJP infection may vary; there may be a rather nonspecific ground-glass or acute pneumonitis-like change or centrilobular nodules (Fig 2a and 2b).3 This can make it difficult to discriminate PJP infection from an acute exacerbation of ILD (AEILD) (see later) or from changes due to other infections (eg viral pneumonitis) on computed tomography (CT) appearance alone. Relevant microbiological samples need to be sent early in the hospital admission (Table 2) and cases should be discussed with the microbiology team as soon as possible, where appropriate.

High-resolution computed tomography (HRCT) images showing variety in the appearance of the lung parenchyma in confirmed Pneumocystis jirovecii (PJP) infection and ground glass-change in an acute exacerbation of idiopathic pulmonary fibrosis. a) A patient with confirmed PJP infection showing changes of bilateral ‘crazy paving’ and extensive ground-glass opacity spreading out from centre to peripheries.3 b) A patient with confirmed PJP infection showing changes of centrilobular nodules in both lungs. The variable appearances of PJP infection on chest X-ray and HRCT can make it hard sometimes to discriminate from other causes of breathlessness in an ILD patient including an acute exacerbation of interstitial lung disease or infection. c) A patient with idiopathic pulmonary fibrosis pre-exacerbation showing reticular change (green arrow) and honeycombing (yellow arrow) in the subpleural area of the right lung base.7 d) A patient during acute exacerbation of idiopathic pulmonary fibrosis showing typical diffuse ground-glass opacities in both lungs (blue arrows).7
In the immunosuppressed patient with ILD, broad spectrum antibiotics may be needed initially with subsequent treatment then targeted depending on culture results and microbiology advice (eg anti-PJP treatment or anti-fungal therapy if fungal infection is suspected). Due to the underlying immunosuppression, a longer course of antibiotics may be required. Microbiological sampling of the airways using bronchoscopy is often difficult and potentially dangerous in acutely hypoxaemic patients but may be appropriate later.
Heart failure, acute pulmonary embolism and lung cancer in the patient with ILD
Distinguishing pulmonary oedema from ILD on a chest X-ray can sometimes be challenging in the patient presenting acutely with breathlessness. This may be particularly so when, as is often the case in the acutely ill patient, the film is technically suboptimal: portable, antero-posterior (AP), rotated and sometimes supine. The presence of the classic radiological features of pulmonary oedema on chest X-ray, including upper lobe pulmonary venous distention, peri-hilar (batwing) shadowing, bilateral pleural effusions and Kerley B lines may be difficult to determine. Where uncertainty exists, reviewing all previous chest radiology for the patient may help. This should include trying to obtain chest X-rays and scans that may have been done elsewhere. Some questions to ask are: ‘Was ILD present before, and if so, how did it appear then and how does this compare with the chest X-ray appearances now?’ Other information to consider in deciding includes a history of ischaemic heart disease or of previous heart failure, previous echocardiography and, of great importance, careful bedside clinical assessment. If uncertainty still exists after this, a trial of intravenous diuretic, provided the blood pressure and renal function permit, with careful assessment of the response, may be reasonable.
Pulmonary hypertension (PH) secondary to ILD is a frequent comorbidity and of concern because its presence is a strong predictor of mortality.4 Patients can develop physiologically compromising right ventricular dysfunction and present with sudden breathlessness. Standard therapies for right heart failure apply, including optimising oxygen therapy to reduce pulmonary pressures and diuretics.
Patients with ILD, particularly those with moderate or severe disease with associated relative immobility, are often at increased risk of venous thromboembolism (VTE). An IPF diagnosis, in particular, confers an approximate two-fold increase in VTE risk. This is because IPF is itself a pro-coagulant condition and because patients with IPF are often older and less mobile with multiple comorbidities.5 CT pulmonary angiography (CTPA) is the investigation of choice because ventilation/perfusion scans are often difficult to interpret in the setting of background fibrosis.
Tip
If CTPA is requested in an acutely breathless patient with ILD, request in addition some HRCT slices. HRCT slices are the best images to permit more accurate evaluation of any ILD pattern present. This may be of particular importance if this is a ‘de novo’ presentation of the patient with their ILD. It is also important to ensure that the patient is adequately oxygenated during the scan and able to undertake it, since it will involve having to lay flat for a short time. If the patient is very breathless, the images may be degraded by movement artefact and therefore may be difficult to interpret.
Patients with IPF are often male, older and have a significant smoking history. Therefore, there is an increased risk of developing lung cancer, amounting to 7%-20%. Adenocarcinoma and squamous-cell carcinoma of the lung are the most frequent histologically and are usually located in the peripheral lung zones where fibrotic changes are predominant. They may, therefore, be difficult to detect, particularly if evolving slowly. The exact driving mechanisms remain unclear but there are multiple common genetic, molecular and cellular processes that link lung fibrosis with lung cancer.6
Acute exacerbations of ILD
An AE-ILD is a clinical diagnosis of exclusion. It has been defined as clinically significant respiratory deterioration of less than 1 month's duration with new radiological abnormalities of diffuse bilateral ground-glass opacification (Fig 2c and 2d) without other clinical causes, such as heart failure or fluid overload, to cause the CT changes.7,8 Additional symptoms can include cough, increased sputum production, fever and flu-like symptoms, confusingly similar to infection. Fig 3 outlines a diagnostic approach to AE-ILD.9

Diagnostic approach to acute deterioration in interstitial lung disease.14 CT = computed tomography; GGO = ground-glass opacity; ILD = interstitial lung disease. Reproduced with permission from Leuschner G, Behr J. Acute exacerbation in interstitial lung disease. Front Med (Lausanne) 2017;4:176.
Acute exacerbations can occur in many types of ILD but more frequently in those with fibrotic patterns on imaging (for example, IPF, fibrotic rheumatoid arthritis-related ILD and fibrotic hypersensitivity pneumonitis (eg fibrotic bird fancier's lung)).10–12
Due to its higher prevalence compared with other ILDs, the majority of AEs seen will likely be in patients with IPF. AEs in IPF have a particularly poor outcome with an in-hospital mortality of greater than 50% and high post-event morbidity.8,13 It is uncertain what triggers them. There may be some unidentified intrinsic factor that causes more rapid disease progression or they may simply represent a response to external factors (such as infection, aspiration, pulmonary embolism or post-anaesthesia for surgery).9
Deciding on treatment for an AE-ILD is difficult due to a lack of clinical trial evidence. Treatment options that should be considered for an AE due to IPF will be different from those for a non-IPF ILD, particularly where the underlying ILD is of an inflammatory nature (eg in a CTD-ILD).
In the past, for AE-IPF, high-dose corticosteroids +/− pulsed immunosuppressive regimens were used, although this was based on low-quality evidence. However, a recent phase III clinical trial (EXAFIP) has shown that the addition of cyclophosphamide to high-dose corticosteroids in the treatment of AE-IPF is potentially harmful, with increased 3-month mortality in those receiving both drugs.14 The authors advise against the use of intravenous cyclophosphamide in this patient group.
For patients with IPF, this study sits consistently with other retrospective reviews and with the PANTHER study, which found that, in more stable patients with IPF, longer-term use of dual immunosuppression (steroids plus azathioprine) is also harmful.15 The jury remains out as to whether a high dose, low dose or ‘no dose’ of steroids is effective at changing the poor outcome of an AE in IPF. One recent study, by Jang et al, has provided data from a retrospective review of AE-ILDs suggesting that low-dose steroids (<1 mg/kg) are preferable in IPF-AE but that for non-IPF-AE higher-dose steroids may be more effective.13
Mounting evidence is pushing clinicians away from an aggressive immunosuppressive approach in AE-IPF. The authors favour a low-dose steroid approach of 30-40 mg/day for 10-14 days until further research data are available. This level of treatment is sometimes indicated anyway for comorbid conditions (such as COPD) that can also contribute to worsening symptoms in some patients with IPF.
There is less evidence to guide management in non-IPF fibrotic ILDs. Good clinical judgement is needed to select the best management approach, including an assessment of ‘how much’ of the patient's disease is clearly fibrotic versus inflammatory, as there can often be some overlap but with one pattern predominating.
The treatment of an AE in an inflammatory ILD is different. Here aggressive dual immunosuppression is more likely to have an impact on improving the outcome of the AE event.
Antibiotics are almost always indicated, at least initially, in the treatment of an AE for any type of ILD. Microbiologically proven bacterial infection during AEs occur at a rate of approximately 20%-25%. Viral infection is found in 5%-10%. PJP infection is more common in patients with ILD on IS drugs (16%) than those with ILD who are not on IS drugs (<3%).13
In summary, treatment of AE-ILD remains difficult due to a lack of high-quality evidence. Clinicians should consider broad spectrum antibiotics or treatment for other specific infections identified in all exacerbating patients, at least initially, particularly in those who are immunosuppressed. A low-dose steroid approach, without intravenous pulsed or oral additional IS drugs is probably the best initial management plan with the current state of knowledge for IPF AEs.
However, high-dose immunosuppressive regimens should be strongly considered for all AEs where the underlying ILD pattern is inflammatory. Before commencing drugs like cyclophosphamide, a respiratory opinion, preferably from someone with ILD expertise, if available, should be sought. Discussion with the respiratory team is also preferred before committing to high-dose intravenous steroids. Now with the wider use of virtual ILD MDTs (since COVID-19), access to discuss difficult cases with the regional ILD specialist centres should be much easier.
Other management strategies in the acutely hypoxaemic patient with ILD presenting on the acute take
Oxygen therapy
It is important to optimise oxygen therapy early. This will often help relieve the significant breathlessness that patients with ILD experience. The longer the patient remains hypoxaemic, the harder they must work to maintain their oxygen saturations, and the more fatigued and distressed they will become. Clinicians should aim for target saturations of 94%-98% (preferably closer to 98% than 94%) if in type I RF. The causes of breathlessness and hypoxaemia in patients with ILD are usually diffusion impairment, V/Q mismatch and reduced pulmonary compliance (stiff lungs). Their peak inspiratory flows during an exacerbation will often be very high indeed and may not be satisfied by using conventional maximum ward level oxygen flow rates of 15 L/min given via a non-rebreathe oxygen mask with reservoir bag.
If available, some patients may gain comfort and symptom relief from high-flow humidified oxygen delivered via nasal cannula (HFNC). These provide oxygen flow rates up to 60 L/min using an air/oxygen blend allowing delivery of a fraction of inspired oxygen (FiO2) that may range from 21% to 100%. Using cannula rather than a facemask also facilitates speaking, eating and drinking.
Non-invasive ventilation in patients with ILD
A full review of the utility of non-invasive ventilation (NIV) support strategies for use in respiratory failure in ILD is outside the scope of this article. However, there are broadly two types of NIV support. Bi-level NIV is used for type II hypercapnic respiratory failure (eg in COPD exacerbations), sometimes also called bi-level positive airway pressure support (BiPAP), has no role in the type I hypoxaemic respiratory failure usually found in ILD (because, by definition, the carbon dioxide levels are normal or low). The other from of NIV support is continuous positive airway pressure (CPAP), which may be delivered via either a nasal, oro-nasal or full-face mask or helmet. The evidence base to support the routine use of CPAP in this setting is currently lacking. It can, however, be considered in individual cases, trialled for patient compliance and usefulness after appropriate assessment by the respiratory team. In the authors' experience and from the literature, the degree of acute dyspnoea in these patients is often such as to prevent them settling comfortably on CPAP and the failure rate is often high.16
Invasive mechanical ventilation
Invasive mechanical ventilation (IMV) is generally not appropriate for patients with advanced IPF, nor for those with other advanced fibrotic ILD, due to very poor outcomes: estimated in-hospital mortality of 90%.8,16 The exception might be the patient with very early IPF or other fibrotic ILD with a good performance status. Each case needs to be assessed based on its own merits in conjunction with intensive care colleagues. Ideally, where possible, patients should have had discussions in outpatient clinics with their respiratory teams prior to any hospitalisation to decide on appropriate ceilings of care.
In inflammatory ILDs, the patients are generally younger and more likely to respond to high-dose IS treatment. IMV should be considered where the chance of a good outcome is suspected. The perception that all AEs of ILD do badly is not correct. A small number of patients can, with the right treatment and support, make a good recovery with a decent quality of life post-event.
Palliative care and symptom management
Patients with ILD often have a high burden of symptoms, particularly breathlessness. Symptom control is very important and at every stage should be assessed and treated. Early palliative care team involvement should be considered, not only in those with advanced ILD but also in those who are suffering but are fully expected to survive. Prompt recognition that patients are approaching the end of life is important and appropriate palliative medications should be used in such circumstances. Symptom management should be a constant adjunct to disease focused care.
- © Royal College of Physicians 2021. All rights reserved.








{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- ABSTRACT
- Key points
- Introduction
- Management of the hypoxaemic patient with ILD presenting to the acute medical take
- Establishing the cause and managing the respiratory deterioration
- Heart failure, acute pulmonary embolism and lung cancer in the patient with ILD
- Acute exacerbations of ILD
- Other management strategies in the acutely hypoxaemic patient with ILD presenting on the acute take
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.