
Abstract
New thrombocytopenia may be associated with a variety of conditions and diagnosis can be challenging. Presentation can vary from life-threatening bleeding or thrombosis to an incidental finding in an asymptomatic patient. New thrombocytopenia requires urgent investigation. Investigations are mainly guided by findings from the clinical history, physical examination, full blood count and blood film analysis. Aside from the actively bleeding patient, rare but life-threatening causes of thrombocytopenia must be identified early as they require urgent treatment. These include thrombotic thrombocytopenic purpura, disseminated intravascular coagulation, suspicion of new acute promyelocytic leukaemia, and vaccine-induced prothrombotic immune thrombocytopenia. Here, we discuss how to approach a patient with new thrombocytopenia, along with key differentials not to be missed.
Key points
In new thrombocytopenia, a repeat full blood count with a citrate- or heparin-coated tube should be sent to exclude pseudothrombocytopenia.
Clinical history, examination, full blood count and blood film analysis are key in narrowing down the many differentials and guiding further investigations.
Thrombocytopenia may be associated with thrombosis, bleeding or both.
Thrombotic thrombocytopenic purpura is a medical emergency with over 90% mortality if untreated, and should be considered in patients presenting with thrombocytopenia and microangiopathic haemolytic anaemia with no clear cause.
Where there is suspicion of new acute promyelocytic leukaemia, retinoic acid treatment should be started urgently with vigorous platelet and clotting factor support.
Introduction
Thrombocytopenia is defined as a platelet count of <150 × 109/L, although patients with a platelet count >50 × 109/L are usually asymptomatic.1 Severe spontaneous bleeding is rare in thrombocytopenia. It is more common when the platelet count is <20 × 109/L, and particularly when <10 × 109/L.
Low platelets may result from reduced production in the bone marrow, increased destruction in the circulation (due to coagulopathic consumption, auto-antibodies, vasculopathy or inflammation), haemodilution or splenic sequestration. Table 1 summarises the common causes of thrombocytopenia.
Differential diagnoses for thrombocytopenia
Initial approach and clinical assessment
In a patient with a new thrombocytopenia, a repeat full blood count along with a citrated or heparinised blood sample should be taken both to confirm thrombocytopenia and to exclude pseudothrombocytopenia caused by artefactual clumping.1 Correct assessment is essential to ensure suitable management of the patient. The initial approach should involve clinical history, examination, full blood count and blood film analysis.
The clinical history aims to establish the cause of the thrombocytopenia. This includes asking about recent infections (viral infections including HIV status), drug/vaccination history (antibiotics/antimalarials/heparin), travel history (malaria / dengue fever), diet (B12/folate deficiencies), past medical history (autoimmune/inflammatory diseases), pregnancy status and alcohol intake, along with establishing any features in keeping with malignancy (night sweats / weight loss / bony pain etc). Bleeding history is mainly helpful in establishing the clinical course and chronicity rather than determining the diagnosis.
Physical examination should involve looking for signs associated with thrombocytopenia including joint, soft tissue or mucocutaneous bleeding, retinal haemorrhage, petechiae, ecchymoses, or skin necrosis. Examining for hepatosplenomegaly, bony tenderness, lymphadenopathy, joint deformities, jaundice, central neurological signs and evidence of liver disease can be useful in establishing the diagnosis.
Examination of the full blood count will identify both the extent of the thrombocytopenia and the potential presence of other cytopenias. Drug-related causes should always be considered. More targeted investigations will be guided by the findings made.
Thrombocytopenia in pregnancy
Platelet count is often lower in pregnancy, partly due to haemodilution and partly from platelet activation. Platelet count drops by roughly 10% in the third trimester.2 This is termed gestational thrombocytopenia (GT). The thrombocytopenia is often mild, however, GT remains a diagnosis of exclusion. In GT, the platelet count should resolve spontaneously within 1–2 months after delivery. Primary immune thrombocytopenia (ITP; described later) is the most common cause of isolated thrombocytopenia in the first to mid-second trimesters.1 Other causes include pre-eclampsia and more rarely HELLP (haemolysis, elevated liver enzymes and low platelets) syndrome. HELLP syndrome is an obstetric and medical emergency.
Primary immune thrombocytopenia
Primary ITP is characterised by an isolated thrombocytopenia. It is an acquired immune disorder more commonly seen in young women and is a diagnosis of exclusion.3 Presentation is varied, some patients present with bleeding, whereas others will be asymptomatic or have minimal bruising or petechial rash. In children, ITP often occurs following an acute viral illness and resolves spontaneously in almost two-thirds. However, in adults, preceding factors are often not identifiable and the course is more chronic. In cases of severe symptomatic ITP, the platelet count can typically be improved using intravenous immunoglobulin accompanied by high-dose steroids. Thrombopoietin agonists and/or immunosuppressive treatments are used for steroid-refractory ITP. Prophylactic platelet transfusions are not recommended in ITP.4
Thrombotic thrombocytopenic purpura
TTP should be treated as a medical emergency. Although rare, TTP is associated with a very high mortality of over 90% if untreated.5 Early plasma exchange reduces mortality rates to 10%–20%. Plasma exchange should be started immediately, ideally within 4–8 hours in a patient presenting with a microangiopathic haemolytic anaemia (MAHA) and thrombocytopenia of no clear cause.
TTP is caused by a deficiency of the von Willebrand factor (VWF) cleaving protein, ADAMTS13. This results in multimers of VWF that cause platelet aggregation particularly in the brain, kidneys and heart. MAHA results from mechanical fragmentation of erythrocytes when passing through semi-occluded vessels (Fig 1).
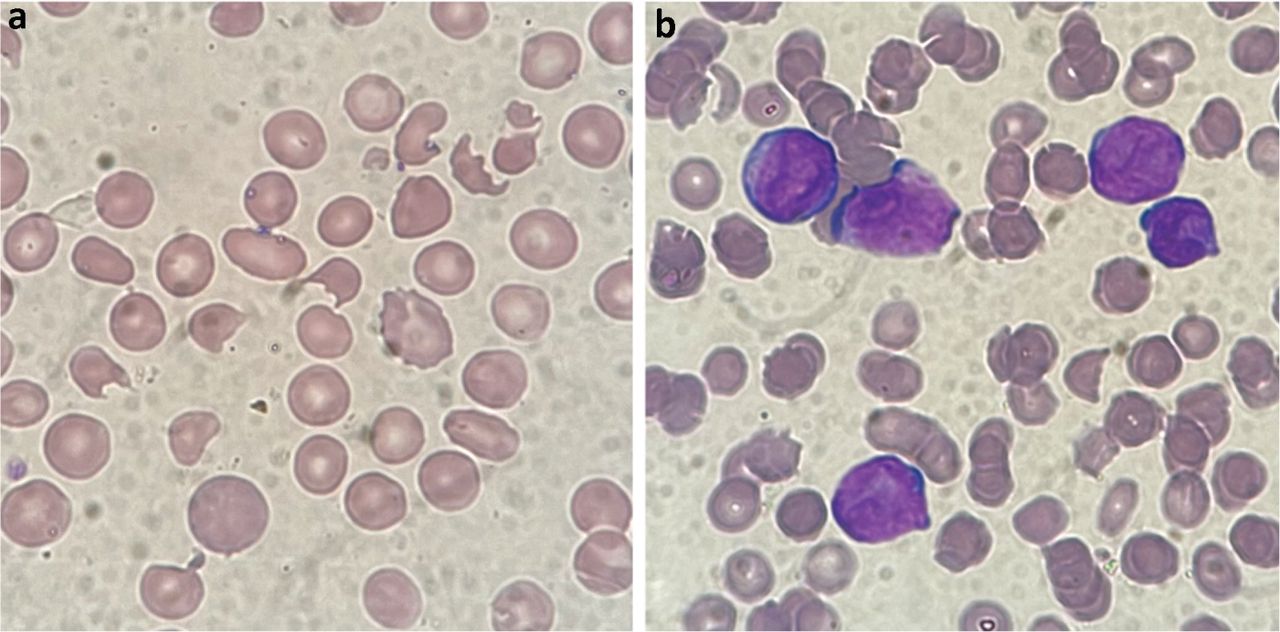
Blood films. a) Red cell fragmentation obtained from a patient presenting with renal failure and confusion secondary to thrombotic thrombocytopenic purpura. b) Thrombocytopenia and features of disseminated intravascular coagulation obtained from a patient presenting with bleeding secondary to acute promyelocytic leukaemia.
Platelet consumption in thrombi causes a marked thrombocytopenia of typically <30 × 109/L at presentation.5 Presentation may be secondary to multi-organ thrombotic events. Neurological symptoms are often flitting and include confusion, paresis, headaches, fluctuating consciousness or coma. Ischaemic events may also cause renal impairment, chest pain or abdominal pain. Other presenting features may include fever and jaundice. The diagnosis is based on clinical history, examination and blood film analysis. An ADAMTS13 assay helps to confirm diagnosis and is used to monitor the disease course.
Haemolytic uraemic syndrome
Haemolytic uraemic syndrome (HUS) is characterised by a triad of thrombocytopenia, MAHA and acute renal failure.6 There may be multi-organ involvement, causing diagnostic overlap with TTP. Typical HUS is commonly associated with verotoxin-producing Escherichia coli 0157:H7, resulting in bloody diarrhoea. It is most common in children and management is supportive.
Atypical HUS (aHUS) is rare but life threatening. It is associated with multi-organ involvement and requires urgent plasma exchange. Distinguishing between TTP and aHUS can be difficult. Renal failure is mainly seen in aHUS. Disruption of the complement system with the monoclonal antibody eculizumab has transformed patient outcomes.7
Disseminated intravascular coagulation
Causes of disseminated intravascular coagulation (DIC) are varied. It is characterised by the aberrant activation of coagulation pathways, microvascular thrombi, and subsequent depletion of coagulation factors and platelets.8 Triggers include sepsis, severe infection, trauma, toxins and some malignancies. Management involves treatment of the underlying disorder, along with supportive management of the coagulopathy. In critically ill non-bleeding patients, venous thromboembolism prophylaxis is recommended while a therapeutic heparin dose should be considered where thrombosis predominates.8 Bloods typically show low platelets, low fibrinogen with a raised D-dimer and prolonged clotting tests.
Acute promyelocytic leukaemia (APML) may present with DIC (Fig 1). Where there is clinical suspicion of APML, patients should be managed as a medical emergency. To prevent early deaths, retinoic acid should be started immediately, along with vigorous correction of the coagulopathy.9
Heparin-induced thrombocytopenia
Heparin-induced thrombocytopenia (HIT) typically occurs between days 5 and 10 after heparin exposure. It is rare beyond day 15 but may occur much sooner (ie before day 5) in patients who have received heparin in the previous 3 months, due to previously formed antibodies.10 HIT is caused by immunoglobulin (Ig) G antibodies against a complex of platelet factor 4 (PF4) and heparin.11 Thrombosis occurs via IgG/PF4/heparin complexes binding to and activating platelets. All patients receiving any heparin should have a baseline platelet count. This typically falls by >50% in HIT.10 In spite of the low platelet count, bleeding is uncommon and the main risk in HIT is thrombosis (both arterial and venous). Half of patients have associated thrombosis at presentation.12 In patients without thrombosis at presentation, they have a 50% risk of developing thrombosis if the heparin is not stopped.12 The risk of HIT in medical patients receiving unfractionated heparin is approximately 0.8%, with a much lower risk in patients receiving low-molecular weight heparin.13,14
The ‘4Ts’ score is used to assess the pre-test probability of HIT (Table 2). A low score means HIT can be excluded. Where the score is not low, heparin must be stopped and an alternative anticoagulant, at therapeutic dose, should be used to reduce the immediate thrombotic risk. Both danaparoid and argatroban are appropriate alternatives and can be switched to warfarin or an oral anti-Xa inhibitor such as apixaban upon platelet count recovery.
The ‘4Ts’ score
Vaccine-induced prothrombotic immune thrombocytopenia
Vaccine-induced prothrombotic immune thrombocytopenia (VIPIT; otherwise known as VITT) is a new and extremely rare post-vaccination thrombotic and embolic syndrome following the vaccination against SARS-CoV-2. It is associated with the AstraZeneca and Janssen vaccinations (both adenoviral-based vector vaccines).15 VIPIT has been defined according to five criteria (Box 1).16 Where all five criteria are met, the diagnosis is deemed ‘definite’. The clinical presentation is similar to HIT. The syndrome typically presents between 5 and 30 days post-vaccination, with a median platelet count at presentation of approximately 50 × 109/L. Cerebral venous sinus thrombosis appears to be the most common site of thrombosis.16 Other common sites include lower limb deep vein thromboses and pulmonary embolisms. It is caused by IgG antibodies that recognise PF4 bound to platelets.17 This results in platelet activation and thromboembolic events, and differs from HIT, where the antibodies are heparin-dependent but not in VIPIT. Current management involves intravenous immunoglobulin and non-heparin anticoagulation.
Criteria for a diagnosis of vaccine-induced prothrombotic immune thrombocytopenia (VIPIT)
Conclusion
Differentials for acute thrombocytopenia are highly varied. Clinical history, examination and blood film analysis play a fundamental role in determining the cause. Rare causes that constitute medical emergencies requiring urgent treatment include TTP, aHUS, APML and VIPIT.
- © Royal College of Physicians 2022. All rights reserved.








{kind=link}
Jump to section
- Article
- Abstract
- Key points
- Introduction
- Initial approach and clinical assessment
- Thrombocytopenia in pregnancy
- Primary immune thrombocytopenia
- Thrombotic thrombocytopenic purpura
- Haemolytic uraemic syndrome
- Disseminated intravascular coagulation
- Heparin-induced thrombocytopenia
- Vaccine-induced prothrombotic immune thrombocytopenia
- Conclusion
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.