
Abstract
Sickle cell disease is a common inherited disorder that is characterised by chronic haemolysis and vaso-occlusive episodes, resulting in severe pain and end-organ damage. The most frequent acute manifestation of sickle cell disease is a painful vaso-occlusive crisis, which can, in some cases, develop into a sickle chest crisis: a life-threatening complication of sickle cell disease that requires early recognition and prompt intervention to prevent progressive respiratory failure. In addition to the acute complications, patients with sickle cell disease are also at risk of a number of chronic complications that require multidisciplinary specialist input.
Key points
Patients with sickle cell disease are usually hyposplenic and should be considered to be immunocompromised.
Chest crisis is a life-threatening acute complication of sickle cell disease and may follow the onset of a painful crisis.
Clinical markers of severity in chest crisis include rising respiratory rate, worsening hypoxia, thrombocytopenia, progressive anaemia, multi-lobar involvement on chest X-ray and neurological impairment.
Patients should have access to care at specialist haemoglobinopathy centres. Expert input is required to prevent end-organ damage and optimise life expectancy.
Individual and structural discrimination against patients with sickle cell disease is regrettably common in the UK and is associated with worse patient outcomes.
Introduction
Sickle cell disease (SCD) originates in areas with high prevalence of falciparum malaria due to the selective advantage conferred by the heterozygous state. African and African-Caribbean populations have the highest incidence, with sickle phenotypes also observed in Mediterranean, Indian and Middle Eastern communities.1,2 Haemoglobin (Hb) S results from a valine to glutamic acid substitution at position 6 of the beta globin chain. SCD arises in individuals who are homozygous for HbS, or in those with a compound mutation, such as HbSC disease (Fig 1; Box 1)1,2. Sickle cell carriers (HbAS) are typically asymptomatic but may experience vaso-occlusive crises when extremely hypoxic.1
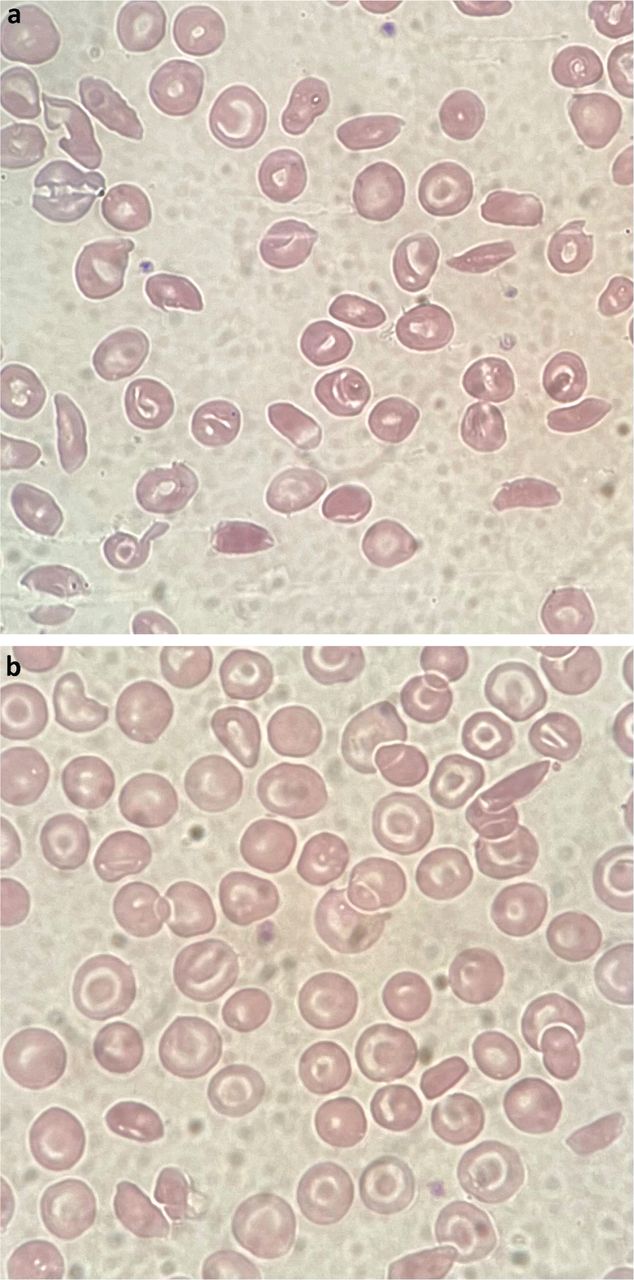
Photomicrography showing red cell changes. a) Haemoglobin SS disease with numerous sickle cells. b) Haemoglobin SC disease with target and sickle cells.
Genotypes associated with sickle cell disease
When deoxygenated, HbS forms insoluble, rod-like structures that aggregate and deform the red cell membrane. This results in chronic haemolytic anaemia and episodes of vaso-occlusion due to aggregation of sickled red cells, leading to pain, tissue hypoxia and end-organ damage.2 The mechanisms surrounding vaso-occlusion are complex, involving mechanical obstruction due to rigid sickled erythrocytes, cellular adherence to vascular endothelium, nitric oxide-induced vasospasm and inflammatory acute phase reactions.1,2
SCD is variable in its phenotype, partly due to concurrent mutations (notably genotypes resulting in raised fetal haemoglobin (HbF)).3 SCD manifests with various acute presentations and also has long-term consequences (Table 1).
Complications of sickle cell disease
Management of a painful crisis
Case 1
A 23-year-old man with SCD presented with a 3-day history of pain in both legs, his lower back and his right shoulder. He reported that he had not been able to control his pain at home with paracetamol, ibuprofen and oral oxycodone. He had a new productive cough.
Acute veno-occlusive crisis (VOC) is a common emergency presentation. Precipitants include dehydration, hypoxia, hypothermia or infection.2 Management hinges on adequate pain control with paracetamol, non-steroidal anti-inflammatory drugs (NSAIDS; if tolerated) and opioids.2 Patients may have a personalised management plan. Patients should be kept warm, well hydrated and given oxygen if saturations are outside the patient's target range.2
Individuals with HbSS are usually hyposplenic due to auto-infarction of the spleen in childhood.2 Careful assessment and low threshold for antimicrobial therapy is required. Penicillin V prophylaxis should be held and restarted on antimicrobial completion. Thromboprophylaxis is important: SCD increases the risk of venous thromboembolism. The local haematology team should be contacted and any planned blood transfusions discussed.
Many patients face difficulties accessing timely pain relief when seeking emergency care for painful crises, and perceived discrimination in this setting is associated with a negative impact on care.4 The No One's Listening report, an All-Party Parliamentary Group inquiry published in November 2021, found serious failings in the treatment received by patients with SCD in the UK. Misconceptions of patients as ‘difficult’ or ‘opiate seekers’ were common, and some patients reported incidents of overt racism.5
Sickle chest crisis
Case 1 continued
The patient was started on paracetamol, naproxen, oxycodone modified-release and pro re nata OxyNorm. He was given intravenous fluids and antibiotics for lower respiratory tract infection as per local trust guidelines. After 2 days, he reported some new chest pain and shortness of breath. His oxygen saturations were 89% on air. A chest X-ray showed new left basal consolidation.
Acute chest crisis is a life-threatening complication of SCD. The definition of a sickle chest crisis is an acute illness characterised by fever and/or respiratory symptoms associated with pulmonary infiltrate on a chest X-ray.6 Thus, a sickle chest crisis encompasses a broad range of underlying pathologies. Chest crisis can arise spontaneously or, in half of cases, follow a VOC. Low oxygen tension increases red cell sickling and vaso-occlusion of pulmonary capillaries, worsening hypoxia and promoting further sickling. Opiate narcosis causing respiratory depression can contribute, as can uncontrolled rib pain leading to hypoventilation. Opiate requirements and toxicity, respiratory rate, and fluid balance should, therefore, be frequently reassessed in all patients.7 Incentive spirometry can prevent hypoventilation and atelectasis.8
Hypoxic adults need an arterial blood gas to establish partial pressure of oxygen (PaO2). A PaO2 <8 kPA or patients in respiratory distress with peripheral saturations <85% on air should trigger intensive care review.8 Untreated, chest crisis can result in progressive respiratory compromise and can be fatal. Predictors of multiorgan failure in chest crisis include thrombocytopenia, falling Hb, increasing respiratory rate, worsening hypoxia, multi-lobar involvement on chest X-ray and neurological compromise.8 As infection can trigger a chest crisis, then blood and sputum cultures, and nasopharyngeal aspirate for respiratory viruses should be taken.7
All patients need an early crossmatch: top up / exchange transfusions can be lifesaving.8 Other aspects of treatment include antibiotics, careful fluid balance, oxygen and early consideration of escalation to intensive care.9
Longer-term management considerations
Input from a haemoglobinopathy specialist improves outcomes. A multidisciplinary approach aids diagnosis and management of end-organ damage. Patients with SCD require a holistic approach that recognises the impact of chronic pain and ill-health on education and employment, and may benefit from input from allied health professionals (such as psychologists, occupational and physiotherapists, and social workers).
Long-term management of SCD is focused on prevention of infection and end-organ damage. Lifelong penicillin V and vaccination against meningococcus, pneumococcus and Haemophilus influenzae B aim to prevent infection with encapsulated organisms. SARS-CoV-2 and annual influenza vaccination should be encouraged, and folic acid replacement is required. Hydroxycarbamide reduces rates of VOC, chest crisis and need for transfusion, and is effective and well tolerated from infancy.10,11 Hydroxycarbamide may lower mortality.12 Current guidance is to offer hydroxycarbamide from 9 months of age regardless of disease severity.12 The mechanism of action is multifactorial and includes increased production of HbF.3
Regular top-up or exchange transfusions may be beneficial for some individuals.1 Women of childbearing age should be counselled on contraception and ideally reviewed by an obstetric specialist prior to conceiving. Medications may need to be reviewed for teratogenicity and shared haematology/obstetrics antenatal care is required.1
New developments
Several new agents are on the horizon. Crizanlizumab, a P-selectin targeted monoclonal antibody (mAb), has recently been approved for the prevention of VOC in individuals aged over 16 years with SCD. Voxelotor is an oral HbS polymerisation inhibitor that reduces haemolysis. L-glutamine reduces hospital admissions, chest crisis and veno-occlusive events.1,2 Neither are currently approved by the National Institute for Health and Care Excellence.
Haematopoietic stem cell transplant can be curative in the <20% of individuals with a matched donor, but it has risks. Finally, there is considerable interest in the potential for gene therapy, with various approaches including restoration of normal HbA function, increasing HbF expression or inducing expression of modified ‘anti-sickling’ haemoglobin.2,13
- © Royal College of Physicians 2022. All rights reserved.








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.