
Abstract
Acute leukaemia is characterised by uncontrolled expansion of immature leukocytes, either myeloid or lymphoid progenitors, leading to acute myeloid leukaemia (AML) and acute lymphoid leukaemia (ALL), respectively. If left untreated, it is life-threatening and can lead to death within weeks. When acute leukaemia is suspected, urgent haematology input should be sought. Appropriate investigations are needed promptly to confirm diagnosis and start treatment. A multidisciplinary approach is vital to ensure appropriate management.
Key points
Acute leukaemia should be considered in any patient with significant leukocytosis, isolated cytopenia, pancytopenia or where blasts are present on the blood film.
Where acute leukaemia is suspected, urgent haematology input should be sought.
The two main types of acute leukaemia are acute myeloid leukaemia (AML) and acute lymphoblastic leukaemia (ALL).
AML is the most common acute leukaemia in adults, while ALL is the most common childhood leukaemia.
Patients with very high white cell counts (hyperleukocytosis) are at risk of leukostasis. This is a medical emergency and typically presents with respiratory or neurological distress.
Introduction
During normal haematopoiesis, haematopoietic stem cells (HSCs) differentiate into mature blood cells via progenitor populations, of which there are two lineages, one involving common lymphoid progenitor (CLP) cells and the other common myeloid progenitor (CMP) cells. HSCs may acquire genetic mutations that transform them into leukaemic stem cells (LSCs) which give rise to acute leukaemia. Acute leukaemia is subdivided into acute myeloid leukaemia (AML) and acute lymphoid leukaemia (ALL). They are characterised by uncontrolled expansion of immature malignant blood cells, which proliferate rapidly, resulting in failure of normal blood formation, termed haematopoiesis. Patients often present with vague non-specific symptoms, which can obscure the diagnosis in the initial stages. However, if left untreated, acute leukaemia is invariably fatal.1
Acute myeloid leukaemia
Introduction
Acute myeloid leukaemia (AML) is characterised by uncontrolled clonal expansion of myeloid progenitors (blasts) in the peripheral blood and bone marrow (BM), where they accumulate and impair the production of healthy haematopoietic cells.2,3 It remains a therapeutic challenge due to its aggressiveness and genetic/phenotypic heterogeneity.4,5 It can arise as a de novo malignancy in previously healthy individuals, secondary to other haematological disorders such as myelodysplastic syndrome, or following previous treatment with cytotoxic agents.6
Diagnosis
The full blood count (FBC) typically shows BM failure (manifesting as anaemia, neutropenia and thrombocytopenia). Leukocytosis is often observed, due to circulating blasts; however, the patient may also present with leukopenia. Confirmation of the diagnosis involves a BM biopsy and generally the presence of more than 20% blasts by microscopic morphological analysis of aspirate and trephine samples. Microscopic findings are usually corroborated with immunophenotyping, specifically CD34 and CD117 expression, which usually correlates with the morphological blast percentage. Other phenotypic markers are used to confirm myeloid lineage such as CD13, CD15 and CD33.
Treatment
For those patients who are previously fit and healthy, without any other significant medical conditions, and are typically below 70 years of age, the management of AML usually involves intensive treatment with curative intent. Induction treatment using a regimen of daunorubicin and cytarabine chemotherapy as an inpatient can result in morphological remission in 80% of adults. However, this will not achieve long-term cure and further consolidation treatment is required, based on the patient's risk stratification performed on the diagnostic BM sample. Low- and standard-risk patients have consolidation chemotherapy, whereas high-risk patients may undergo an allogeneic stem cell transplant. Novel targeted agents such as midostaurin (an FLT3 inhibitor), gemtuzumab ozogamicin (Mylotarg; an anti-CD33 monoclonal antibody) and Vyxeos (liposomal daunorubicin and cytarabine) are increasingly used according to the results of the initial molecular and cytogenetic analysis. For patients who are not fit for intensive chemotherapy, venetoclax in combination with azacitidine has been shown to improve outcomes, particularly in older patients, where this treatment has rapidly become the standard of care. The treatment for acute promyelocytic leukaemia (APML), an aggressive subtype of AML that commonly presents with severe disseminated intravascular coagulopathy, involves all-trans-retinoic acid (ATRA) and is curable in 90% of patients.1
Prognosis
The long-term disease-free survival (DFS) for AML is only 40% at 6 years.4 Adverse prognostic features include age (>75 years old), high white cell count, unfavourable cytogenetics including complex karyotype, host status (organ dysfunction or a low performance status) and longer duration of antecedent haematologic disorders.7,8 In current clinical practice, patients are routinely risk stratified into ‘good risk’ (presence of t(15;17), t(8;21) and inv(16) translocations), ‘intermediate risk’ (normal karyotype) and ‘poor risk’ (adverse or complex karyotype) categories. Additional prognostic information is gained from analysis of the mutational status of two genes: the FLT3 gene, typically an indication for allografting where possible, and the NPM1 gene, which if FLT3-wildtype often predicts better response to chemotherapy and is associated with a more favourable outcome.9,10
Acute lymphoblastic leukaemia
Introduction
Acute lymphoblastic leukaemia (ALL) can affect both adults and children. It is an aggressive leukaemia of lymphoid precursor cells, either B cell or T cell. The lymphoblasts accumulate in the BM, suppressing normal haematopoiesis. They can also accumulate in other organs such as the lymph nodes, spleen, liver, central nervous system (CNS) and testicles. It is hypothesised that ALL in children develops in utero (‘first hit’), followed by further postnatal genetic alterations.11
Diagnosis
Similar to AML, the FBC can show either pancytopenia or leukocytosis, with lymphoblasts on the blood film. A BM biopsy and aspirate is routinely performed to assess the extent of BM involvement. A cut-off of ≥20% lymphoblasts in the BM is used to make the diagnosis. Lymphoblasts (either from peripheral blood, BM aspirate or tissue biopsy) are sent for immunophenotyping to identify the lineage-specific antigens as well as cytogenetics. B lymphoblasts are typically positive for CD19, CD10, CD34 and nuclear terminal deoxynucleotide transferase (TdT), whereas T lymphoblasts commonly express CD2, CD3, CD7, CD34 and TdT.12
Treatment
Intensive chemotherapy with curative intent involves induction chemotherapy followed by consolidation chemotherapy. Agents used include anthracyclines, cytarabine, vincristine and asparaginase. Regimens are targeted towards preventing infiltration of the CNS via intrathecal chemotherapy and high–dose methotrexate. Tyrosine kinase-inhibitors may be used in ALL patients who are Philadelphia-positive (Ph+), which is more common in adults and confers a worse prognosis. Consolidation chemotherapy is followed by a maintenance phase with 6-mercaptopurine and methotrexate and can last 2 years. Novel therapies such a blinatumomab and inotuzumab may be used at relapse. Stem cell transplantation is generally reserved for adults with high-risk disease and children with relapse.13
Prognosis
Cure rates are high in children but poor in adults, probably due to the different cytogenetic changes observed in these age groups. Factors associated with worse prognosis include older age and CNS involvement. Cytogenetic analysis, karyotype and morphology are also important. Patients with Philadelphia chromosome t(9;22) ALL have a poor prognosis and represent more than 30% of adult cases. Similarly, chromosomal abnormalities such as t(4;11), deletion of chromosome 7, and trisomy 8 have also been shown to confer a poor prognosis.
Acute leukaemia should be suspected in any patient with significant leukocytosis, isolated cytopenia, pancytopenia or blasts on the blood film. If an acute leukaemia is suspected, haematology should be involved urgently. A thorough history is vital as patients often present with vague symptoms. Clinicians should specifically ask about symptoms of anaemia (fatigue, breathlessness, chest pains and dizziness), thrombocytopenia (bruising and bleeding), neutropenia (infection) and systemic signs (weight loss, sweats and fevers). Past medical history and general health is important, especially any evidence of pre-existing cancer or previous chemotherapy. Clinical examination should look for signs of pallor, cachexia, gum hypertrophy, splenomegaly or hepatomegaly and for signs of infection.
Box 1 and Fig 1 describe an illustrative case history.
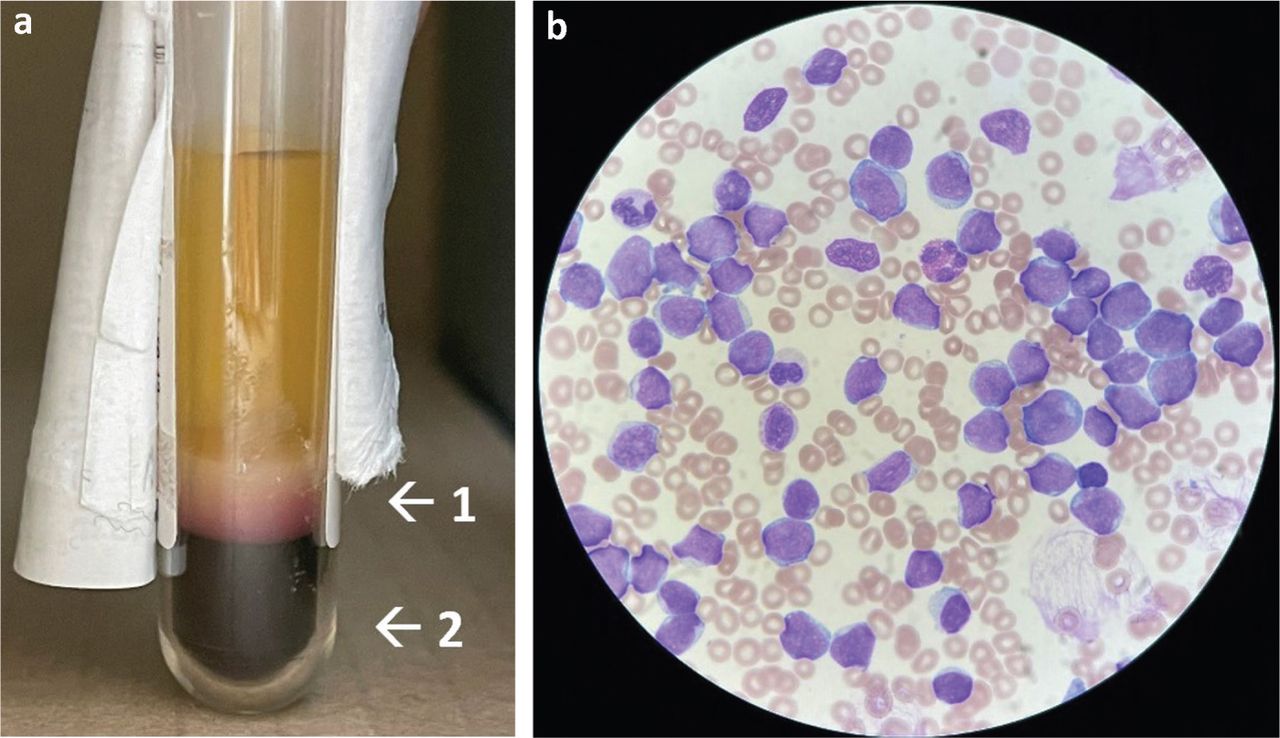
Blood test findings indicating acute lymphoid leukaemia. a) The patient's blood sample was centrifuged showing thick buffy coat layer (arrow 1) and reduced haematocrit (arrow 2), indicating hyperleukocytosis and anaemia respectively. b) Patient's blood film confirmed large numbers of leukaemic blasts with high nuclear to cytoplasmic ratio and lymphoblastic morphology.
Case history
Emergency presentations
Patients with acute leukaemia may present to hospital as an emergency. Below are the most common presentations that clinicians should be aware of.
Bone marrow failure
Bone marrow failure is indicated by the presence of anaemia, leukopenia/neutropenia and/or thrombocytopenia.
Anaemia is defined as a red blood cell count of below 130 g/L for men or 115 g/L for women. In acute leukaemia, due to bone marrow failure, the anaemia tends to be significant, and a transfusion is usually indicated to keep haemoglobin above 80 g/L.
Neutropenia is defined as mild (1–1.5 × 109/L), moderate (0.5–1 × 109/L) or severe (below 0.5 × 109/L). In the case of fever and neutropenia, prompt antibiotic treatment is critical as neutropenic sepsis can be life-threatening if left untreated.
Thrombocytopenia: platelet transfusion is usually indicated where platelets are below 10 × 109/L, platelets are below 20 × 109/L in the presence of infection, or platelets are below 30 × 109/L with bleeding.
Leukostasis
Leukostasis is a medical emergency, where the blast cell count is extremely elevated, often >100 × 109/L, with microvasculature blockage causing reduced tissue perfusion. Urgent treatment with leukapheresis is needed to lower the white cell count. Signs and symptoms of leukostasis include shortness of breath and hypoxia, and neurological impairment such as visual disturbance, headache, dizziness, confusion and risk of intracranial haemorrhage.
Disseminated intravascular coagulation
Disseminated intravascular coagulation (DIC) can occur in all types of leukaemia, but more frequently in acute promyelocytic leukaemia (APML). Laboratory findings include prolonged prothrombin time (PT) and activated partial thromboplastin time (APTT), thrombocytopenia, low fibrinogen, raised D-dimers and red cell fragments on blood film. Urgent treatment with retinoic acid and vigorous clotting factor and platelet support is essential if APML is suspected.
Tumour lysis syndrome
This is caused by rapid leukemic cell death following the onset of chemotherapy. It is characterised by hyperphosphataemia, hypocalcaemia, hyperuricaemia, hyperkalaemia and renal insufficiency.
- © Royal College of Physicians 2022. All rights reserved.








{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.