
Case history
A 52-year-old man presented with a two-month history of pain in the right ankle, metacarpophalangeal (MCP) joints bilaterally, left shoulder and lower back along with pins and needles in both hands. Past medical history was negative for any medical illness and there was no relevant family history. He smoked 10 cigarettes per day and his alcohol consumption was 14 units of beer per week. He was a manual worker. On examination he had bony thickening at the second and third MCP joints, diminished sensation over median nerve distribution, negative Tinel's sign and positive Phalen's test of median nerves bilaterally. Movements in the lumbar spine were uncomfortable but full. Neurological examination of legs was normal. The working diagnosis was osteoarthritis with bilateral carpel tunnel syndrome (CTS).
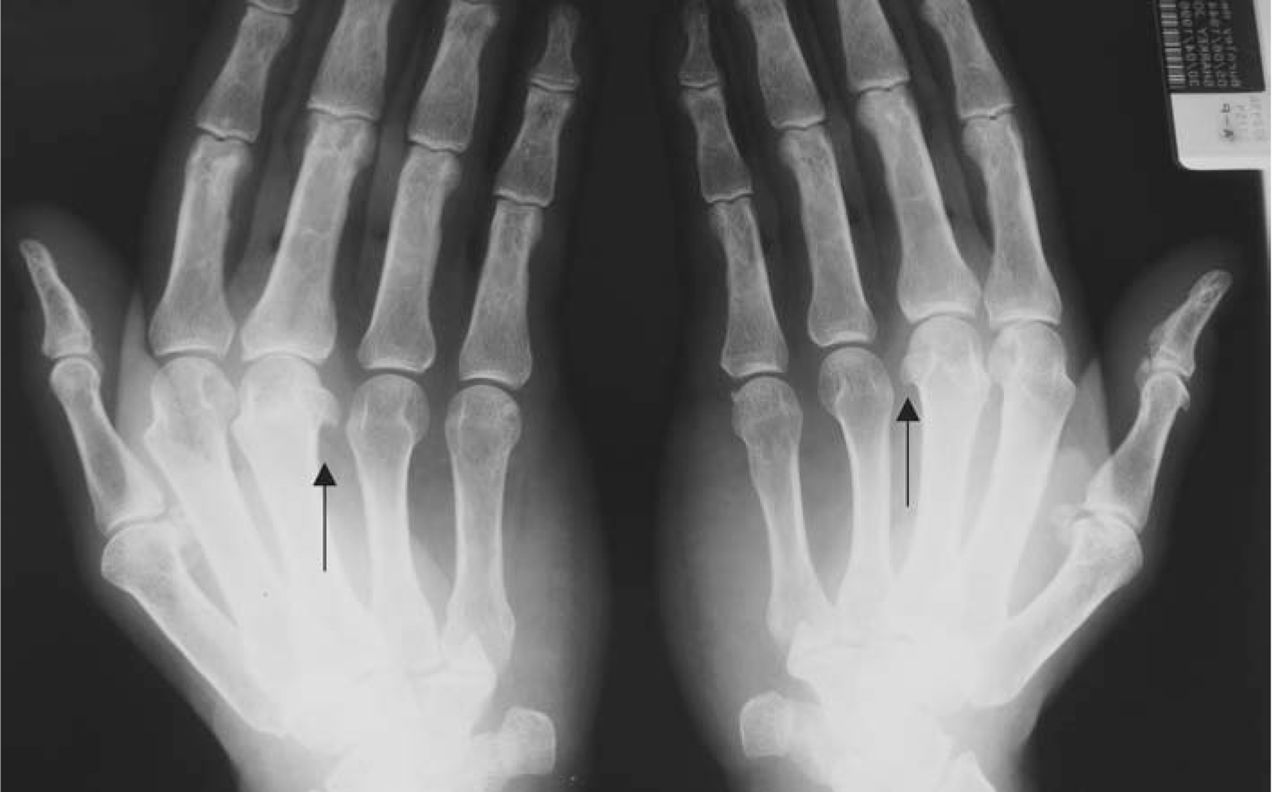
Initial investigations revealed normal full blood count, erythrocyte sedimentation rate, glucose tolerance, liver, renal and thyroid functions. Rheumatoid factor was negative. X-ray of hands revealed osteoarthritic changes and hook shaped osteophytes in the second and third MCP joints (Fig 1). Chest X-ray and electrocardiograph were normal. Nerve conduction revealed bilateral CTS.

X-ray of hands showing hook shaped osteophytes.
Further investigations revealed the serum iron to be 41 umol/l, total iron binding capacity (TIBC) 51%; ferritin 597.6 ugm/l; transferrin saturation 80.39% and testosterone 22.2 IU/l (normal). Genetic testing revealed the patient to be homozygous for C282Y gene, which confirmed the diagnosis of HH. His liver biopsy was normal and he has been receiving regular venesections since the diagnosis. The latest bloods show normal liver functions, iron 31 umol/l, TIBC 50.8 umol/l, ferritin 47 ugm/l, and transferrin saturation at 61.2%.
After careful counselling, further screening of his seven siblings revealed three brothers were positive for C282Y homozygous while the two other brothers and two sisters were positive for heterozygous gene (C282Y) and are being followed up with regular measurement of iron status.
Discussion
Hereditary haemochromatosis is the most common inherited metabolic disorder in the western world with autosomal recessive patterns.2 The gene involved lies close to human leucocyte antigen region of chromosome 6 and prevalence varies from 1:2000 to 1:200 in various population of North European origin.1 In the UK, over 90% of the HH patients are homozygous for C282Y mutation and another 4% are compound heterozygotes (C282Y/H63D).1
Since the description of the human hemochromatosis protein (the HFE gene) mutations by Feder and colleagues genotypes from patients with HH have been determined in many countries.3 Of patients satisfying the criteria for HH, 60–100% have been found to be homozygous for the C282Y mutation, about 4% appear to be compound heterozygotes (C282Y, H63D) and about 6.7% are heterozygous for C282Y or H63D mutation.1 HH is a disorder of iron absorption and storage with tissue damage due to iron deposition.2 Iron deposition in the synovium does not correlate with radiological evidence of arthropathy or joint destruction.4
The clinical presentation is variable and not restricted to cirrhosis, diabetes and skin pigmentation (bronze diabetes). Patients are predominantly men (ratio of 9:1) and usually present between the age of 40 and 60 years with lethargy, weakness and sleep disturbance with diabetes or gonadal failure. These subtle symptoms, however, can be overlooked.5
Niederau and colleagues reported that life expectancy was normal for patients who began treatment before the development of diabetes or cirrhosis and that hepatic fibrosis could be improved with iron removal.3 Once cirrhosis develops life expectancy shortens along with the risk of carcinoma despite venesection.1
Transferrin saturation of more than 55% for men and 50% for premenopausal women suggests iron accumulation. This should be followed by a fasting sample to confirm elevated saturation levels. Although raised transferrin saturation provides an early indication of iron accumulation, it is not necessarily raised in young people who are homozygous for the haemochromatosis gene and there are other causes of raised levels. After confirmation of raised transferrin saturation, serum ferritin concentration should be checked. These do not exceed the upper limit of normal until liver iron concentrations are elevated and then they rise disproportionately with the degree of liver damage. False positives include acute and chronic inflammatory conditions and cholestasis.1
Genetic testing is not influenced by any of the above factors. However, it is not certain that the majority of people who are homozygous for C282Y mutation will develop the clinical condition. People with both C282Y and H63D mutation may also accumulate iron but the risk of clinical haemochromatosis is much less. In the UK, about 5% of patients lack mutations of the HFE gene and currently only biochemical tests can detect iron overload in this group.1 Liver biopsy has no role in diagnosis but helps in establishing the degree of liver fibrosis. A suggested approach for liver biopsy is raised transferrin saturation, ferritin concentration >1,000 mcgm/l and evidence of liver damage as revealed by hepatomegaly or raised aspartate transaminase. No treatment is necessary for those with normal values for transferrin saturation and ferritin. For homozygous C282Y mutation without iron accumulation, it is reasonable to monitor iron status yearly.1
The treatment for haemochromatosis is regular venesection to lower the body's iron stores. Venesection is performed initially every week until the ferritin concentration is less than 20 mcg/l and transferrin saturation is less than 16%. The venesection rate is reduced if anaemia develops, or ferritin concentration drops below 50 mcg/l, but maintenance venesection is required so that transferrin saturation is kept below 50% and the ferritin level is maintained to be less than 50 mcgm/l.1
Arthropathy in haemochromatosis is quite common and can present without other features of the disease, as described above. It characteristically involves the second and third MCP joints, with stiffness. The arthropathy is degenerative rather than inflammatory and can lead to extensive joint destruction with attacks of pseudogout.5 The symptoms of arthralgia, diabetes mellitus and hypogonadism may improve with treatment in some patients.1 Although there is no measurement of iron status that will define a point at which signs and symptoms become irreversible.1 Morbidity among first degree family members of C282Y homozygous probands is higher than in the normal population.6 As there is increased risk of serious illness among siblings of HH patients with increasing exposure to HH gene, especially diabetes, arthritis, hepatoma,7 it is extremely important to have proper counselling. In one study, the siblings were motivated for screening tests due to the seriousness of HH but barriers, including lack of symptoms and belief that the condition is rare, are common.8 A literature search has failed to show similar numbers (eight) in one family having positive screening.
Conclusion
If the typical radiological manifestations are detected early long-term complications, such as diabetes, cirrhosis and hepatocellular carcinomas, in patients and siblings can be prevented.
- © 2009 Royal College of Physicians








{kind=link}
Related Articles
Cited By...
- No citing articles found.