
Abstract
The role of drotrecogin alfa (activated) (DAA) in severe sepsis remains controversial and clinicians are unsure whether or not to treat their patients with DAA. In response to a request from the European Medicines Agency, Eli Lilly will sponsor a new placebo-controlled trial and history suggests the results will be subject to great scrutiny. An academic steering committee will oversee the conduct of the study and will write the study manuscripts. The steering committee intends that the study will be conducted with the maximum possible transparency; this includes publication of the study protocol and a memorandum of understanding which delineates the role of the sponsor. The trial has the potential to provide clinicians with valuable data but patients will only benefit if clinicians have confidence in the conduct, analysis and reporting of the trial. This special article describes the process by which the trial was developed, major decisions regarding trial design, and plans for independent analysis, interpretation and reporting of the data.
Similar content being viewed by others


Introduction
The incidence of severe sepsis is increasing and mortality rates remain high. Whilst the case fatality rate has decreased modestly, the overall number of deaths is still increasing [1, 2]. Severe sepsis affects approximately 10 ± 4% of ICU patients, and around 100 ± 50 persons per 100,000 population [2–10]. The burden of disease and economic impact of sepsis are considerable and probably comparable to that of ischaemic heart disease [4].
As new treatments for patients with severe sepsis have proven remarkably elusive [11], the publication of the PROWESS trial reporting that drotrecogin alfa (activated) (DAA) reduced mortality was met with considerable enthusiasm [3–10, 12, 13]. Subsequently, much of the initial enthusiasm has waned and opinion over DAA has become polarized [14, 15]. At its annual review of DAA in February 2007, the European Medicines Agency (EMEA) stated: “Taking all the available data together, the Committee for Medicinal Products for Human Use (CHMP) considered that the benefit/risk balance of Xigris required additional clarification and that there was a need for another clinical trial to further prove the efficacy of Xigris in the target population….therefore a placebo-controlled study in patients … with severe sepsis and documented organ failure (e.g. MOD or vasopressor-dependent septic shock) when treated within a strictly defined time window, should be performed to assert the benefit/risk profile of Xigris.” [16].
Eli Lilly and Company (Lilly) agreed to sponsor the study and subsequently assembled an academic steering committee to advise on the design the trial and interpret its results. This article, written by the academic steering committee, describes the scientific and academic background to the study and is accompanied by publication of the full study protocol as an on-line supplement. Additionally, it presents the memorandum of understanding (MoU) delineating the relationship between the steering committee and the sponsor. The MoU recounts the processes by which the steering committee, the independent academic statistical centre (ASC) and the data monitoring committee (DMC) were established and formalizes the respective roles of the sponsor and these committees in the conduct of the trial. The processes and relationships described in the MoU are intended to ensure that the study will be designed, conducted, and analysed to the highest possible scientific standards, and be reported transparently. The primary goal for the SC is to supervise the design, conduct and analysis of a clinical trial that seeks to answer the crucial question: “Does DAA reduce mortality in patients with severe septic shock?”
Background
The PROWESS study, the first phase III placebo-controlled randomised controlled trial of DAA, was stopped early following a second planned interim analysis when 1,690 of the planned 2,280 subjects had been enrolled. The trial reported an absolute reduction in 28-day all-cause mortality of 6.1% (a relative risk reduction [RRR] of 19.4%) [12].
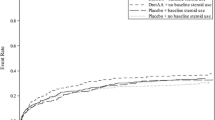
Although the overall trial result was statistically significant, an FDA advisory committee noted several concerns; in particular, approximately half way through recruitment the protocol had been amended and the drug manufacturing process changed and treatment with DAA appeared more beneficial after these changes (Fig. 1).

Cumulative mortality rate over time in the PROWESS trial. Solid black lines, drotrecogin alfa (activated) group; solid grey lines, placebo group. The amended version of the protocol was introduced at Line A, first interim analysis occurred at Line B and the second interim analysis at Line C. (Reproduced from Critical Care Medicine 2004;32(12):2388 with permission)
Additionally, the FDA advisory committee evaluated the treatment effect within subgroups and concluded that there was no apparent benefit in subjects at lower risk of death as indicated by lower APACHE II scores or in those patient with single organ dysfunction at baseline [17].
The FDA advisory committee split 10:10 on whether DAA should be approved for clinical use [18]. The FDA received statistical advice that the variable treatment effect observed over the course of the trial would be expected in approximately 8% of clinical trials and that the trial results did not appear to be due to the mid-trial protocol amendment or the change in the drug manufacturing process [19]. On 21 November 2001, the FDA licensed DAA but only for the treatment of patients with severe sepsis and a high risk of death (as determined, for example, by the APACHE II score), and requested additional studies in children and in adult patients with severe sepsis and a lower risk of death [20]. In Europe, the European Medicines Agency (EMEA) authorized DAA to be used for the treatment of adult patients with severe sepsis with multiple organ failure when added to best standard care; this authorization was subject to annual review [16].
The additional trials requested by the FDA
Two additional placebo-controlled trials have been conducted. The ADDRESS study in adult patients with severe sepsis who were at a lower risk of death was stopped early after 2,640 participants had been enrolled (Table 1) [21]. The independent DMC estimated the likelihood of demonstrating a reduction in 28-day mortality to be less than 5%. Within the ADDRESS Trial mortality data were available for 321 subjects (12.3%) who had a baseline APACHE II score of 25 or more, the subgroup that appeared to benefit in PROWESS; within this subgroup, 28-day mortality was 29.5% in those assigned to DAA versus 24.7% in those assigned placebo, in-hospital mortality rates were 32.3 and 32.7%, respectively [21]. Similarly, there was no reduction in mortality in the subgroup of patients within ADDRESS who had multiple organ dysfunction at baseline [21].
Between November, 2002 and April, 2005, the RESOLVE trial randomly assigned children with sepsis-induced cardiovascular and respiratory failure to receive placebo or DAA [22]. The study was stopped after the second interim analysis when 477 participants had been enrolled when the independent DMC advised that the likelihood of demonstrating a beneficial effect of DAA was low (Table 1) [22].
Evidence of harm
Whilst the efficacy of DAA is now the subject of much debate, the evidence that DAA increases the risk of clinically significant bleeding is consistent across trials (Table 2). In ENHANCE, an open label clinical evaluation where DAA was given to patients who satisfied the entry criteria for the PROWESS Study, the serious bleeding rate was 5.5%, somewhat higher than that seen in the subjects who received DAA in the PROWESS study [23]. Outside clinical trials off-label use may be common and the incidence of bleeding may be increased. In one clinical series, the incidence of bleeding was 10.9% and patients who received DAA and suffered bleeding were more likely to die than those who received DAA and did not bleed [24]; in the absence of a control group assessment of how much bleeding is attributable to the administration of DAA is not possible.
Expert commentary and the implications for patients
Results of these studies and the split vote of the FDA advisory committee generated considerable controversy and calls for a confirmatory trial were made as early as 2002 [25]. The results of the ADDRESS and RESOLVE studies resulted in further calls for another placebo controlled trial [26–30]. The recent Cochrane review and meta-analysis also questioned whether DAA should be used to treat patients with severe sepsis and a high risk of death, describing the evidence of efficacy as “very weak” [30].
Furthermore, all three placebo-controlled trials were stopped early; the PROWESS study was stopped for efficacy, an approach that tends to overestimate the beneficial effects of treatments [31, 32].
With expert opinion divided on the balance of the benefits and risks of treating patients with DAA, clinical use has varied markedly between individual hospitals and clinicians with many eligible patients not receiving the drug [33–35]. The variable use of DAA has serious implications for patients; if DAA does reduce the risk of death then many patients are being denied an effective treatment and dying unnecessarily; conversely, if DAA does not reduce the risk of death, other patients are being treated with an ineffective and expensive agent that may cause clinically significant bleeding.
Can an industry-sponsored trial resolve this controversy?
It seems clear that the current controversy can only be resolved by additional randomised placebo-controlled trials. As industry sponsored trials tend to report more favourable results than independent research [36, 37], and as some clinicians will be concerned by allegations that Lilly funded the Surviving Sepsis Campaign and used it to market DAA [38, 39], the most credible trial for all concerned would not be funded or conducted by Lilly. Ideally, the trial would be conducted by independent academic clinical trialists who would be responsible for the data, conduct the statistical analysis, interpret the results and write the study manuscripts. One such investigator initiated and trial of DAA, funded by the French government, is planned to run in parallel with the Lilly-sponsored PROWESS-SHOCK trial [40]; both trials will focus on patients with persistent septic shock. These two trials in combination, be they positive or negative, have great potential to settle this controversy. As the PROWESS-SHOCK trial is being sponsored by Lilly at the behest of the EMEA, it will fall short of some commentators’, and our, vision of the most credible trial. With this in mind, the steering committee has sought to develop trial processes that can give clinicians and patients confidence in the trial results; we describe below the process by which the trial was developed, some of the major decisions made regarding trial design, and plans for an independent analysis and reporting of the data.
The PROWESS-SHOCK Trial
Organizational structure and role of the study sponsor (Lilly)
Lilly has appointed a contract research organization (Parexel International) to be responsible for the day-to-day conduct of the study at each participating centre. At Lilly’s invitation, Drs Taylor Thompson and Marco Ranieri agreed to act as co-principal investigators for PROWESS SHOCK and co-chair an academic steering committee. Additionally, patient recruitment and protocol adherence will be scrutinized by a clinical coordinating centre; the clinical coordinating centre is located at Vanderbilt University and is commercially contracted by Lilly to provide these services. The steering committee is not privy to the terms of the contract but is satisfied that the study design prevents the clinical coordinating centre from introducing bias into the study. The statistical analysis will be performed by both Eli Lilly and an academic statistical centre and the safety of trial participants will be overseen by an independent data monitoring committee (DMC) (see Appendix 1), The members of the DMC will be reimbursed at an hourly rate ($250–$350/h) for participation in conference calls, and face to face meetings addressing data monitoring issues related to the PROWESS-Shock trial. The relationship between Lilly and the study committees is governed by a signed MoU (see text box).
Memorandum of Understanding |
1. The two principal investigators for the PROWESS–SHOCK study, Dr Thompson and Dr Ranieri, were invited to Co-Chair the committee by Eli Lilly and Company (Lilly). The principal investigators selected the other seven members of the steering committee (SC) in cooperation with Lilly. The principal investigators independently approved the members and had the right to veto members proposed by Lilly. Lilly representation in the SC will be limited to three ex officio non-voting members. |
2. The steering committee in collaboration with medical experts from Lilly designed the protocol for PROWESS-SHOCK, after Lilly had come to an initial agreement with the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMEA) to study patients with septic shock. Lilly provided analyses from their sepsis database to assist protocol design and provided input regarding statistical and regulatory aspects of the study. The two principal investigators led the protocol design meetings and approved the final protocol. Lilly did not have the right to veto any steps of this process. |
3. Dr. Ranieri recommended the chair of the Data Monitoring Committee (DMC) and invited Dr. Slutsky to participate. Dr. Slutsky, the two principal investigators, and Lilly recommended members of this committee. Dr. Slutsky approved the final membership of the committee and had the right to veto members proposed by Lilly. |
4. The two principal investigators identified an academic statistical center that (subject to contractual agreement) will be responsible for reviewing and approving the statistical analysis plan, including all of the prospectively defined analyses for efficacy and safety. The plan will be filed with the FDA prior to the data being unblinded and at the same time it will be made freely and publicly available on an academic website. The academic statistical center will be responsible for conducting the primary analysis of the study data, and preparing the main study report for the SC to create the primary manuscript, and any additional analyses of the study data for subsequent manuscripts that in the view of the SC are material to understanding the efficacy and safety of drotrecogin alfa (activated). The academic statistical group will also be responsible for performing any post-hoc analyses requested by the steering committee. The members of the academic statistical center will have unfettered access to the full study database and to the randomisation code at the time the study randomisation code is broken. Eligible members of the academic statistical center will be co-authors on study manuscripts. Statisticians appointed by Lilly will independently conduct the same analyses and any discrepancies will be resolved to the satisfaction of both parties. All documents required for regulatory purposes will be prepared by Lilly. |
5. The monthly SC meetings via teleconference and/or all face-to-face meetings will include an open session with Lilly experts and closed meetings independent of Lilly. The principal investigators are responsible for documenting any discussions that occur during the closed meetings of the SC. |
6. The sponsor has pledged to make public, after publication of the main paper, the final study report for the study which will include the following components: Introduction, Full Protocol, Investigational Plan, Statistical Methods, Disposition of Patients, Protocol Violations, Efficacy Evaluations, Safety Evaluations, and Tables of baseline and on study variables as well as analyses of all outcomes, relevant Figures and Graphics, and Final Conclusions. In the two years following publication of the main paper, the academic statistical group will provide additional analyses of the full database as requested by any member of the public and approved by the SC. The SC will develop procedures for reviewing public requests, judging the merit of these requests for additional analyses, and adjudicating duplicate requests for access to the database. Lilly will not have the right to veto any requests the SC approves. The cost of any additional analysis will be paid by the person or group requesting the analysis. |
7. The two principal investigators are responsible for the main publications from this study. There will be no scientific writer from Eli Lilly and Company for the principal manuscript or subsequent manuscripts approved by the SC. Appropriate co-authors from the sponsor will be included if they meet standard criteria for authorship. However, (a) the two principal investigators and the independent members of the steering committee will be responsible for final approval of the content and conclusions of the manuscripts; (b) the manuscripts will be submitted by the two principal investigators not by Lilly; (c) Lilly will not have the right to veto any steps of this process. |
8. Lilly will remunerate the members of the SC and DMC or their employing institutions for time spent performing committee activities and the amounts paid to each committee member will be stated at any major presentation of study results and in all manuscripts reporting study results that are authored by the SC. Time spent in preparation of manuscripts and lecture materials will not be remunerated by Lilly. Any lecture materials regarding the conduct or results of the study will be clearly identified as either prepared and approved by the Steering Committee or prepared by Lilly independent of the steering committee. |
9. Following publication of the primary manuscript SC and DMC members will have an absolute right to make public comment in any medium should they have concerns about the design, conduct or analysis of the study, or the interpretation or presentation of the study data. |
Study design and goals
PROWESS-SHOCK will be a concealed, randomised placebo-controlled trial in which patients, care-givers, data collectors, statisticians, the study steering committee, and the clinical coordinating centre will be blinded. Whilst mindful of the results of the PROWESS, ADDRESS and RESOLVE studies, and Lilly’s obligations to drug registration agencies, the primary goal of the trial is to provide clinicians with robust evidence regarding the efficacy and safety of DAA in a clearly defined and clinically important patient population.
Proposed time-lines for the trial are to start recruitment in March 2008, complete recruitment in July 2010 and have 28-day mortality data available for public dissemination by the end of 2010.
Choice and rationale for study population (Protocol Sect. 4.1)
The trial must satisfy regulatory requirements but equally it must provide clinicians with a clearly identifiable patient population to which the study results, be they positive or negative, can be applied. The EMEA obliged Lilly to complete a trial in patients “with severe sepsis and documented organ failure (e.g. multiple organ dysfunction [MOD] or vasopressor dependent septic shock)”. The steering committee has elected to make persistent vasopressor-dependent septic shock (defined as the continuous requirement for a vasopressor above a minimum threshold dose for at least four hours) the key inclusion criterion as such patients are clearly identifiable in clinical practice and appeared to benefit in the PROWESS trial. They also represent the majority of patients that clinicians who use DAA treat with the drug [41]. Standard definitions will be used to identify patients with severe sepsis [42]. In addition, treatment with the vasopressor must be continued through the time required to obtain informed consent and for the study pharmacist to be ready to prepare the study drug; patients’ whose septic shock has resolved during this time will not be eligible for randomisation. Furthermore, patients must have one further clinical sign consistent with hypoperfusion (Table 3). The patients within the PROWESS study who most closely satisfied these inclusion criteria had a 28-day mortality of 29.3% for those assigned to DAA and 37.6% for those assigned placebo (RR 0.78, 95% CI 0.63–0.96; Lilly—data on file). The inclusion criteria are listed in Table 3; based on these inclusion criteria, we estimate a placebo mortality rate of 35%. Thus a study of 1,500 patients will provide 80% power to detect a relative risk reduction of 20% with an α of 0.05.
As a further safeguard to ensure as far as possible that only eligible patients enter the trial, all patients will be screened by a clinical coordinating centre prior to randomisation. The staff of the clinical coordinating centre (CCC) will review a checklist of inclusion and exclusion criteria with site investigators or coordinators and must approve randomisation. Reasons for advising against randomisation will be recorded and reviewed monthly by the academic steering committee. As should be the case in any randomised controlled trial, treatment allocation will be concealed with local investigators and CCC staff blinded to the randomisation sequence so that decisions to include or exclude patients will be made with no knowledge of the patient’s potential treatment group. To minimize the risk of a centre effect and reduce the effect of an imbalance in concomitant treatments between the two groups, randomisation will be stratified by centre. As far as is practical, the use of concomitant therapies that are likely to influence the outcome from septic shock will be captured in the case report form and reported.
To reduce the risk of recruitment bias, participating centres conducting competing trials in patients with septic shock will use a predetermined random or sequential selection procedure for deciding which trial a patient enters when a patient is eligible for more than one trial.
Short term and longer term outcomes (Protocol Sect. 6.1)
The primary outcome measure will be all-cause mortality in the intention-to-treat population (all patients randomised regardless of whether they received all or any study treatment) 28-days after randomisation. Survival and quality of life will be assessed for up to 180 days after randomisation. The secondary outcome measures are listed in the protocol.
Adaptive design to allow an increase in sample size if overall mortality is lower than expected (Protocol Sect. 8.1.1)
Intensive care treatment of patients with severe sepsis and septic shock is changing and the case fatality rate is decreasing [1, 2, 43]. Against this background, an accurate prediction of the placebo group mortality is difficult. Therefore, the steering committee has made provision for the study statisticians to calculate the blinded mortality rate when approximately 750 patients have been enrolled, if the overall mortality rate is less than 30% the sample size may be increased by up to 500 subjects.
Rationale for conservative efficacy stopping rule and absence of “futility” stopping rule (Protocol Sect. 8.2.8)
The DMC and the steering committee separately discussed the principles for interim analyses and stopping rules; both committees were in favour of very conservative stopping rules and that the trial should not be stopped for futility. The DMC recommended the final stopping rules for the study and these were accepted by the steering committee.
Two interim analyses are planned; the first after approximately one-third and the second after two-thirds of the planned number of subjects have completed 28 days of follow up. The DMC will be supplied with unblinded data; these data will not be seen by the steering committee or the sponsor. For the first interim analysis, no efficacy stopping rules are planned. We reasoned that only an implausibly large benefit of DAA over placebo would stop the trial after recruitment of 500 subjects and that such an effect should be regarded with great scepticism [44]. In addition, it is highly likely that the number of deaths accrued would fall below that recommended for stopping such a trial [31]. It is clear that seemingly convincing treatment effects observed early in trials may not be sustained when trials are allowed to complete recruitment as planned [32, 44].
As one experienced commentator stated “Decisions on early stopping (or not) need to be based on wise judgments interpreting the totality of available evidence, both in the current trial (considering primary and other efficacy outcomes and safety issues) and in other external evidence (especially from related trials). Accordingly, a statistical stopping boundary is only one useful objective component in an inevitably more challenging decision making process.” [32] Additionally, in the event that the mortality in the DAA group was lower at the first interim analysis, we consider it ethical to continue the trial as the proportion of eligible patients receiving DAA outside the trial is likely to be much lower than the 50% within the trial [24, 33].
For the second interim analysis at 1,000 patients, an efficacy stopping guideline is proposed if DAA is superior to placebo at a p value of ≤0.001 and if at least 250 deaths have occurred [31]. At this point, we reason that such an effect would be compelling and would be sufficient to convince many clinicians to change their practice.
We decided against a formal futility stopping guideline because of the importance to determine with as much certainty as possible whether DAA is ineffective and thus discontinue its use in usual care. However, the independent DMC may recommend the trial be stopped for safety concerns at any time.
A comparison of the PROWESS and PROWESS-SHOCK trials is presented in Table 4.
Safety monitoring (Protocol Sect. 6.2)
Site investigators are responsible for reporting all serious adverse events and any non-serious bleeding or thrombotic events that occur within 28 days of randomisation. After 28 days, only serious adverse events that the site investigator considers to be related to the study drug, the drug delivery system, or a protocol procedure are reported. The DMC will be provided with monthly reports regarding safety data. If after reviewing these reports the DMC has concern regarding the incidence of adverse or serious adverse events in either arm of the study, the DMC can request and will be granted an unscheduled review of unblinded safety data. The DMC will also review unblinded data at all scheduled interim analyses.
Ethics of a placebo-controlled trial of licensed drug in high risk population: identifying study sites with equipoise/uncertainty
There are a number ethical issues and practical challenges in conducting a placebo controlled trial of a licensed drug within the current indication for use and in a vulnerable patient population. Central to their consideration is that the evidence in favour of DAA must now be considered equivocal, numerous respected commentators have called for another such trial [25–30], and currently relatively few patients with severe sepsis receive DAA [24, 33–35]. From an individual patient perspective, any patient for whom the treating clinician considers DAA to be clearly indicated or contra-indicated will be excluded from the trial, thus the trial will only recruit patients where the senior treating clinician has clinical equipoise or substantial uncertainty over the balance of the benefits and risks associated with treatment with DAA. An important criterion for the selection of trial centres is that the local clinicians are uncertain about the benefits and risks of treatment with DAA and consider it ethical to treat eligible patients with a blinded study drug that may be either DAA or placebo.
Another risk to the conduct of the trial is that clinicians may wish to treat patients who are deteriorating whilst receiving study drug with commercially available DAA; this risk will be minimized by selecting centres where clinicians are uncertain about the benefits and risks of treatment with DAA and where clinicians report that this uncertainty has resulted in minimal use of DAA. These aspects were assessed in a questionnaire as part of the participating centre evaluation, the document evaluating potential participating centres is reproduced in Appendix 2. Additionally, the physical properties of DAA mean that it is possible for caregivers to become “unblinded” to treatment allocation and this might theoretically influence the use of concomitant treatments (such as commercially available DAA) or willingness to continue active medical treatment. The use of commercially available DAA and the issuing of “DNR” orders will be monitored during the trial and reported.
Maximizing clinician acceptance of the PROWESS SHOCK results
The controversy over the PROWESS study results has been documented elsewhere [19, 25, 26, 38]. We hope to reduce the risk of similar controversy following the current trial; we created conservative efficacy stopping guidelines to assure a more precise estimate of the overall treatment effect and reduce the concern that trials stopped early overestimate treatment effects [31, 45]. The trial will use an academic clinical coordinating centre to assist investigators in recruiting appropriate patients and in following the protocol precisely. The clinical coordinating centre will serve as a resource for study procedures which should minimize the learning curve and its potential impact on the trial results [46]. Finally, we hope that publishing the trial protocol, the memorandum of understanding governing the relationship between Lilly and the study committees and full disclosure of all prior and present relationships relevant to the trial, together with plans for independent analysis of the data, we will provide clinicians and regulators with sufficient information to allow them to fully evaluate the conduct of the trial and to interpret the results.
Summary
Although the initial report of the PROWESS study suggested a significant breakthrough in the search for treatments for patients with severe sepsis, the subsequent controversy surrounding the conduct of that trial and the subsequent negative trials, have left many clinicians uncertain whether to treat their patients with DAA or not. There have been a number of calls for another placebo controlled trial some of which have specifically called for a trial run by a not-for-profit organisation [24]. Whilst Lilly will sponsor a new trial, the steering committee has put in place processes which it hopes will increase the transparency with which the trial is conducted so that the trial can provide robust evidence that will be acceptable to clinicians. This is an ethical imperative, as only by providing credible evidence and resolving this controversy can we serve the best interests of our patients.
References
Dombrovskiy VY, Martin AA, Sunderram J, Paz HL (2007) Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med 35:1244–1250
Martin GS, Mannino DM, Eaton S, Moss M (2003) The Epidemiology of Sepsis in the United States from 1979 through 2000. New Engl J Med 348:1546–1554
Alberti C, Brun-Buisson C, Burchardi H, Martin C, Goodman S, Artigas A, Sicignano A, Palazzo M, Moreno R, Boulme R, Lepage E, Le Gall R (2002) Epidemiology of sepsis and infection in ICU patients from an international multicentre cohort study. Intensive Care Med 28:108–121
Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR (2001) Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 29:1303–1310
Cheng B, Xie G, Yao S, Wu X, Guo Q, Gu M, Fang Q, Xu Q, Wang D, Jin Y, Yuan S, Wang J, Du Z, Sun Y, Fang X (2007) Epidemiology of severe sepsis in critically ill surgical patients in ten university hospitals in China. Crit Care Med 35:2538–2546
Finfer S, Bellomo R, Lipman J, French C, Dobb G, Myburgh J, the ANZICS CTG Sepsis Investigators (2004) Adult population incidence of severe sepsis in Australian and New Zealand Intensive Care Units. Intensive Care Med 30:589–596
Karlsson S, Varpula M, Ruokonen E, Pettila V, Parviainen I, la-Kokko TI, Kolho E, Rintala EM (2007) Incidence, treatment, and outcome of severe sepsis in ICU-treated adults in Finland: the Finnsepsis study. Intensive Care Med 33:435–443
Linde-Zwirble WT, Angus DC (2004) Severe sepsis epidemiology: sampling, selection, and society. Crit Care 8:222–226
Padkin A, Goldfrad C, Brady A, Young D, Black N, Rowan K (2003) Epidemiology of severe sepsis occurring in the first 24 hrs in intensive care units in England, Wales, and Northern Ireland. Crit Care Med 31:2332–2338
Salvo I, de Cian W, Musicco M, Langer M, Piadena R, Wolfler A, Montani C, Magni E (1995) The Italian SEPSIS study: preliminary results on the incidence and evolution of SIRS, sepsis, severe sepsis and septic shock. Intensive Care Med 21(Suppl 2):S244–S249
Russell JA (2006) Management of Sepsis. N Engl J Med 355:1699–1713
Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ Jr (2001) Efficacy and safety of recombinant human activated protein C for severe sepsis. [comment]. New Engl J Med 344:699–709
Matthay MA (2001) Severe sepsis—a new treatment with both anticoagulant and antiinflammatory properties. N Engl J Med 344:759–762
Mackenzie I, Lever A (2007) Management of sepsis. BMJ 335:929–932
Mackenzie AF, Bartelink AK (2007) Management of sepsis. N Engl J Med 356:1179
European Medicines Agency (2002) Marketing authorization. Drotrecogin alfa (activated). Available from http://www.emea.europa.eu/humandocs/PDFs/EPAR/xigris/247102en7.pdf. Accessed 8 November 2007
Anti-Infective Advisory Committee. (2001) FDA briefing document: drotrecogin alfa (activated) [recombinant human activated protein C (rhAPC)] Xigris, BLA#125029/0. Food and Drug Administration, September 12, 2001. Available from http://www.fda.gov/ohrms/dockets/ac/01/briefing/3797b1.htm. Accessed 11 August 2007
Food and Drug Administration (2001) CDER 2001 meeting documents. Available from http://www.fda.gov/ohrms/dockets/ac/cder01.htm#Anti-Infective. Accessed 11 August 2007
Siegel JP (2002) Assessing the use of activated protein c in the treatment of severe sepsis. N Engl J Med 347:1030–1034
Food and Drug Administration (2002) Approval Letter - Drotrecogin alfa (activated), Xigris. Available from http://www.fda.gov/cder/foi/appletter/2001/droteli112101L.pdf. Accessed 6 September 2007
Abraham E, Laterre PF, Garg R, Levy H, Talwar D, Trzaskoma BL, Francois B, Guy JS, Bruckmann M, Rea-Neto A, Rossaint R, Perrotin D, Sablotzki A, Arkins N, Utterback BG, Macias WL, the Administration of Drotrecogin Alfa (Activated) in Early Stage Severe Sepsis (ADDRESS) Study Group (2005) Drotrecogin Alfa (Activated) for Adults with Severe Sepsis and a Low Risk of Death. N Engl J Med 353:1332–1341
Nadel S, Goldstein B, Williams MD, Dalton H, Peters M, Macias WL, bd-Allah SA, Levy H, Angle R, Wang D, Sundin DP, Giroir B (2007) Drotrecogin alfa (activated) in children with severe sepsis: a multicentre phase III randomised controlled trial. Lancet 369:836–843
Vincent JL, Bernard GR, Beale R, Doig C, Putensen C, Dhainaut JF, Artigas A, Fumagalli R, Macias W, Wright T, Wong K, Sundin DP, Turlo MA, Janes J (2005) Drotrecogin alfa (activated) treatment in severe sepsis from the global open-label trial ENHANCE: further evidence for survival and safety and implications for early treatment. Crit Care Med 33:2266–2277
Bertolini G, Rossi C, Anghileri A, Livigni S, Addis A, Poole D (2007) Use of Drotrecogin alfa (activated) in Italian intensive care units: the results of a nationwide survey. Intensive Care Med 33:426–434
Warren HS, Suffredini AF, Eichacker PQ, Munford RS (2002) Risks and benefits of activated protein C treatment for severe sepsis. N Engl J Med 347:1027–1030
Mackenzie AF (2005) Activated protein C: do more survive? Intensive Care Med 31:1624–1626
Friedrich JO, Adhikari NK, Meade MO (2006) Drotrecogin alfa (activated): does current evidence support treatment for any patients with severe sepsis? Crit Care 10:145
Opal SM (2007) Can we RESOLVE the treatment of sepsis? Lancet 369:803–804
Gardlund B (2006) Activated protein C (XigrisR) treatment in sepsis: a drug in trouble. Acta Anaesthesiol Scand 50:907–910
Marti-Carvajal A, Salanti G, Cardona AF (2007) Human recombinant activated protein C for severe sepsis. Cochrane. Database. Syst Rev. Issue 3. Art. No.: CD004388. DOI: 10.1002/14651858.CD004388.pub2
Mueller PS, Montori VM, Bassler D, Koenig BA, Guyatt GH (2007) Ethical issues in stopping randomized trials early because of apparent benefit. Ann Intern Med 146:878–881
Pocock SJ (2005) When (not) to stop a clinical trial for benefit. JAMA 294:2228–2230
Dombrovskiy V, Martin A, Sunderram J, Paz H (2006) Use of drotrecogin alfa (activated) for severe sepsis in New Jersey acute care hospitals. Am J Health Syst Pharm 63:1151–1156
Muller L, Jaber S, Raillard A, Lefrant J (2008) Use of recombinant human activated protein C in patients with severe sepsis: a French retrospective multicentre study. Intensive Care Med 34:977–979
Schultz MJ, Levi M (2006) Prescription of rh-APC differs substantially among western European countries. Intensive Care Med 32:630–631
Jorgensen AW, Hilden J, Gotzsche PC (2006) Cochrane reviews compared with industry supported meta-analyses and other meta-analyses of the same drugs: systematic review. BMJ 333:782–786
Lexchin J, Bero LA, Djulbegovic B, Clark O (2003) Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 326:1167–1170
Eichacker PQ, Natanson C, Danner RL (2006) Surviving sepsis—practice guidelines, marketing campaigns, and Eli Lilly. N Engl J Med 355:1640–1642
Singer M (2006) The surviving sepsis guidelines: evidence-based or evidence-biased? Crit Care Resusc 8:244–245
University of Versailles (2008) Activated Protein C and Corticosteroids for Human Septic Shock (APROCCHS). Available from http://clinicaltrials.gov/ct2/show/NCT00625209. Accessed 3 January 2008
Higgins TL, Steingrub JS, Tereso GJ, Tidswell MA, McGee WT (2005) Drotrecogin Alfa (Activated) in sepsis: initial experience with patient selection, cost, and clinical outcomes. J Intensive Care Med 20:291–297
Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ (1992) Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. Am College Chest Physicians/Soc Crit Care Med Chest 101:1644–1655
The Australasian Resuscitation in Sepsis Evaluation (ARISE) Investigators, the Australian, New Zealand Intensive Care Society (ANZICS) Adult Patient Database (APD) Management Committee (2007) The outcome of patients with sepsis and septic shock presenting to emergency departments in Australia and New Zealand. Crit Care Resusc 9:8–18
Wheatley K, Clayton D (2003) Be skeptical about unexpected large apparent treatment effects: the case of an MRC AML12 randomization. Control Clin Trials 24:66–70
Montori VM, Devereaux PJ, Adhikari NKJ, Burns KEA, Eggert CH, Briel M, Lacchetti C, Leung TW, Darling E, Bryant DM, Bucher HC, Schunemann HJ, Meade MO, Cook DJ, Erwin PJ, Sood A, Sood R, Lo B, Thompson CA, Zhou Q, Mills E, Guyatt GH (2005) Randomized trials stopped early for benefit: a systematic review. JAMA 294:2203–2209
Macias WL, Vallet B, Bernard GR, Vincent JL, Laterre PF, Nelson DR, Derchak PA, Dhainaut JF (2004) Sources of variability on the estimate of treatment effect in the PROWESS trial: implications for the design and conduct of future studies in severe sepsis. Crit Care Med 32:2385–2391
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is discussed in the editorials available at: doi:10.1007/s00134-008-1274-6; 10.1007/s00134-008-1302-6 and 10.1007/s00134-008-1303-5.
An erratum to this article can be found at http://dx.doi.org/10.1007/s00134-010-2081-4
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendices
Appendix 1
Members of the Data Monitoring Committee
Arthur S. Slutsky (Chair)
St. Michael’s Hospital; University of Toronto, Toronto, Canada
Derek C. Angus
University of Pittsburgh School of Medicine, Pittsburgh, USA
Raymond J. Carroll
Texas A&M University, College Station, USA
Timothy W. Evans
Royal Brompton Hospital and Imperial College London, London, UK
Gordon Guyatt
McMaster University, Hamilton, Canada
Charles Weijer,
University of Western Ontario, London, Canada
Appendix 2
Potential investigator questionnaire
Eli Lilly and Company is sponsoring a placebo controlled study to further define the benefits and safety profile of Xigris®, drotrecogin alfa (activated), in adult patients with persistent septic shock. PAREXEL is the contract research organization for this study.
PAREXEL and Lilly are in the process of selecting qualified investigators and investigative sites in select countries around the world. Please review the protocol highlights and complete and return the questionnaire if you are interested in participating in this study.
Investigator selection will be completed this fall. Patient enrollment will begin in the first quarter of 2008.
Study drug
drotrecogin alfa (activated) or placebo.
Study objectives
The study objectives are to investigate efficacy (28-day all-cause mortality) and safety in adult patients with septic shock that are treated with either drotrecogin alfa (activated) or placebo.
Study design
Patients will be randomly (1:1) assigned to either the drotrecogin alfa (activated) or placebo treatment group.
The study will enroll adult patients who meet the following criteria:
-
(a)
Evidence of an infection and the presence of SIRS
-
(b)
Presence of septic shock
-
The patient must have received ≥ 30 mL/kg intravenous fluid resuscitation
-
The patient must have a continuous requirement for vasopressor support for at least 4 hours at a minimum dose (Norepinephrine ≥ 5 mcg/min, or similar dose of dopamine, phenylephrine, epinephrine, or vasopressin)
-
(c)
At least one clinical sign consistent with hypoperfusion (sepsis-induced)
-
Metabolic acidosis (base deficit ≥ 5.0 mEq/L, venous bicarbonate < 18 mEq/dL or lactate ≥ 2.5 mMol/L)
-
Renal injury (urine output < 0.5 mL/kg/h for 1 hour or a 50% increase in creatinine)
-
Acute hepatic dysfunction (AST or ALT > 500 IU/dL or bilirubin > 2 g/dL).
-
(d)
Study drug infusion must begin within 24 h of septic shock onset (i.e. after initiation of vasopressor therapy) and within 36 h of any non-cardiovascular sepsis-induced organ dysfunction.
If you are not the proper person to review and complete this questionnaire, kindly forward this to the appropriate person at your institution.



Conflict of Interest
A statement by the authors concerning their financial relationship with regard to the subject of this paper is available as Electronic Supplementary Material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 2.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by-nc/2.0/.
About this article
Cite this article
Finfer, S., Ranieri, V.M., Thompson, B.T. et al. Design, conduct, analysis and reporting of a multi-national placebo-controlled trial of activated protein C for persistent septic shock. Intensive Care Med 34, 1935–1947 (2008). https://doi.org/10.1007/s00134-008-1266-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-008-1266-6